氧化石墨烯/聚吡咯插层复合材料的制备和电化学电容性能
2013-09-17门春艳
石 琴 门春艳 李 娟
(新疆大学化学化工学院,乌鲁木齐830046)
1 引言
超级电容器是一种新型的能量存储装置,与蓄电池和传统电容器相比,具有容量大、功率密度大、使用寿命长、经济环保等优点,广泛应用于通信、电动汽车、军事和工业等众多领域.1-3目前,常用的电极材料有:具有高比表面积的碳材料、金属氧化物材料和导电聚合物材料.导电聚合物具有比电容高、电子导电性好和内阻小等优点引起了国内外学者的广泛关注.其中,聚吡咯(PPy)因制备过程简单、成本低、电导率高、电化学可逆性好被普遍认为成最具有应用前景的电极材料之一.4,5但是,单纯的PPy机械性能较差,在反复充放电的过程中,PPy链的体积不断收缩膨胀,分子链的结构被破坏致使其导电性能逐渐减弱,表现出较差的循环性能.6-8为了克服这一缺点,可将PPy与碳材料复合以提高其电化学性能,如:碳纤维、碳纳米管和石墨烯等.
石墨烯具有比表面积大、高导电性、循环稳定性和机械性能好等优点,用于超级电容器电极材料有其独特的优势.9,10但由于石墨烯片层受范德华力的影响容易发生团聚或叠加,11导致其表面积大大降低,同时,也不利于聚合物单体进入层间形成复合材料.然而,氧化石墨烯(GO)是石墨烯的特殊衍生物,其表面有大量的含氧官能团(环氧基、羧基和羟基等),这些亲水基团使GO具有较好的水溶性,能以单层片体的形式分散在水中.12,13因此,GO/PPy复合材料引起部分研究者们的兴趣,如:张海英等12用原位聚合法制备GO/PPy复合物在电流密度为0.5 A·g-1下的比电容为401.5 F·g-1,1200圈循环稳定性测试后,容量保持率为46.4%,有待改善.Chang等14用电化学法合成GO/PPy复合膜,在1 A·g-1下的比电容为289 F·g-1,但未研究其循环性能.
因此,本文选用FeCl3和甲基橙(MO)在水溶液中形成的纤维状络合物(FeCl3-MO)为自降解模板,15采用化学原位聚合法使吡咯单体在GO层间交错聚合,形成插层结构的复合材料,并分析了插层结构的形成过程.此外,进一步地研究了GO/PPy插层复合材料作为超级电容器的电极材料在两种不同水系电解液(1 mol·L-1Na2SO4和1 mol·L-1H2SO4)中的电化学行为,探讨了复合材料的循环稳定性.
2 实验部分
2.1 试剂与仪器
甲基橙(天津市恒兴化学试剂制造有限公司,分析纯),无水三氯化铁(沈阳市新西试剂厂,纯度≥99%),无水硫酸钠(天津市河东区红岩试剂厂,纯度≥99%),浓硫酸(天津市风船化学试剂科技有限公司,纯度为95%-98%),无水乙醇(天津市百世化工有限公司,纯度≥99.7%),丙酮(天津基准化学试剂有限公司,纯度≥99.5%),吡咯(Py)单体(Aladdin化学药品公司,分析纯)重新蒸馏后,在0°C下存放备用.
采用日本Mac M18ce型X射线衍射仪进行XRD测定,实验条件:CuKα辐射 (λ=0.154056 nm),管电压40 kV,管电流100 mA,扫描范围2θ为10°-80°,扫描速率10(°)·min-1;用德国Leo1430VP型扫描电子显微镜和日本Model Hitachi H-600,200 kV透射电镜观察样品的形貌.工作电极在上海辰华仪器有限公司CHI660A电化学工作站做循环伏安、恒流充放电性和交流阻抗测试,在武汉金诺公司Land电池测试仪(Land,CT2001A)上进行恒电流充放电性能测试.
2.2 GO/PPy复合材料的制备
采用改进的Hummers法16制备氧化石墨烯(GO),备用.准确称量20 mg氧化石墨烯置于30 mL去离子水中,超声波分散处理2 h,加入0.049 g甲基橙,超声波分散处理1 h,随后加入0.486 g FeCl3,再超声波分散处理30 min,在搅拌下滴加0.201 g吡咯单体(GO与吡咯单体的质量比为1:10),于0-5°C下静置24 h后,解冻,抽滤得黑色沉淀,用去离子水、丙酮、无水乙醇反复洗涤多次.60°C下真空干燥24 h后取出,充分研磨,即得GO/PPy复合材料.
纯聚吡咯的制备方法同上,不加入氧化石墨烯.
2.3 GO/PPy电极的制备及电化学表征
将1 mg样品分别与0.3 mg乙炔黑和1滴聚四氟乙烯(PTFE)乳液混合均匀制成有一定粘度的糊状,均匀涂覆在石墨电极上.50°C下,干燥过夜.以样品的电极为工作电极,饱和甘汞电极(SCE)、铂电极分别为参比电极和对电极,1 mol·L-1Na2SO4和1 mol·L-1H2SO4为两种电解液形成三电极体系,在CHI660A电化学工作站做循环伏安、首次恒流充放电性和交流阻抗测试,在Land电池测试仪上进行恒流充放电循环性能测试.
3 结果与讨论
3.1 样品的XRD表征
图1为PPy,GO和GO/PPy复合材料的XRD图.从纯PPy的XRD谱图中可以看到,2θ为15°-30°的区域内有一个宽峰,显示PPy是无定形结构.而在GO的XRD谱图中,在2θ为11.28°处出现了GO特征衍射峰,17,18对应于(001)晶面,片层间距约为0.78 nm,远大于石墨的片层间距(0.34 nm),这是由于在GO层上、层间及边缘存在大量的含氧官能团(羧基、羟基及环氧基等),使得GO的层间距增大.GO与PPy复合后,由于复合物中GO含量低,而且在超声波的作用下,间距本就较宽的GO片层进一步剥离开来,导致2θ=11.28°的衍射峰弱到几乎消失并继续向小角度方向移动.此外,GO/PPy复合物在2θ=15°-30°之间出现一个宽峰,与纯PPy相比,峰型变得宽而弱,表明无定形的PPy已成功插层到剥离开的GO层间形成复合物.
3.2 样品的FTIR分析
图2是样品PPy,GO和GO/PPy的红外光谱图.从PPy的谱图可以看出:在1541和1448 cm-1附近的吸收峰为吡咯环的特征峰,分别归属于聚吡咯环的对称伸缩振动和非对称伸缩振动,1301和1031 cm-1附近的吸收峰分别对应于C-N键的伸缩振动和C-H键的面内弯曲振动,886和780 cm-1吸收峰归属于吡咯环的C-H键的面外弯曲振动,在1161 cm-1有较强的吸收峰是PPy掺杂态的特征峰.19在GO的谱图中3420 cm-1处附近有很强的宽吸收峰,应为GO表面含氧官能团中O-H伸缩振动峰,1721和1400 cm-1分别对应于羧基的C=O伸缩振动和C-OH官能团中的O-H变形振动峰,1069 cm-1对应于环氧官能团中C-O伸缩振动吸收峰,此外,在1632 cm-1处有明显的吸收峰可以归因于石墨在氧化的过程中吸收了水分子.20这些特征峰表明氧化石墨烯中存在多种含氧基团(极性基团),如:羧基、羟基、羰基、环氧基等.在GO/PPy复合材料的谱图中,上述PPy和GO的特征峰均存在,峰形相似,而GO谱图中的C=O伸缩振动峰(1721 cm-1)明显变弱且发生红移,与1632 cm-1处的吸收峰叠加后形成复合物谱图中1640 cm-1处的吸收峰,表明GO层间的-COOH与吡咯中的-NH形成了氢键.
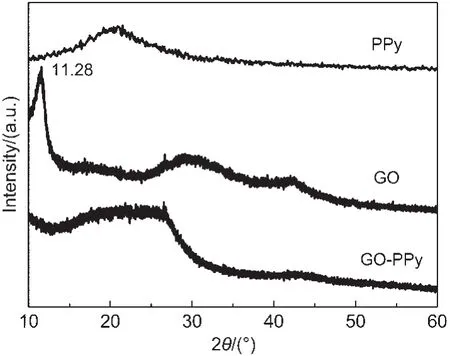
图1 PPy、GO及GO/PPy复合材料的XRD图Fig.1 XRD patterns of PPy,GO,and GO/PPy composite
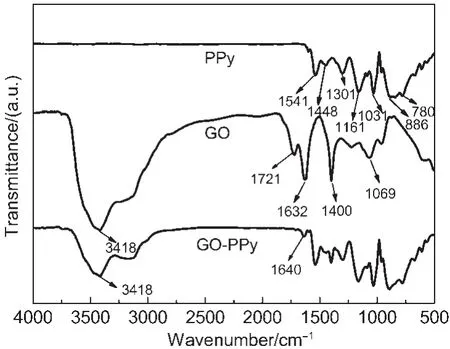
图2 PPy、GO和GO/PPy复合材料的FTIR谱图Fig.2 FTIR spectra of the PPy,GO,and GO/PPy composite
3.3 GO/PPy插层复合材料的微观形貌及形成过程分析
据文献报道,用Hummers法制备的GO通常是单层或数层结构,21,22单层石墨烯表面有一定程度的褶皱,而随着石墨烯层数的增加褶皱程度越来越小.23图3(a,c)为GO、GO/PPy复合物的SEM图,图3(b,d)为GO、GO/PPy复合物的TEM图.从图3a可以看出,GO的形貌为卷曲、褶皱,表面有许多纹络的片层结构,与文献16相符.与PPy复合之后(图3c)可以清楚地看到,复合物的形貌保持了GO的片状结构,片层明显增厚且表面疏松、粗糙、有很多凸起,这是由于PPy在GO层间及表面附着所致.为了更好地分析GO/PPy复合物的结构,我们对样品做了TEM分析.图3b展示了氧化石墨烯典型的片层结构,颜色较浅,但可以看出该氧化石墨烯片是数层堆叠在一起,并非以单层形式存在.从GO/PPy复合材料的TEM(图3d)中可以明显看出图中有深色和浅色区域,浅色区域仍具有片层形貌为GO,深色区域为交织的PPy链状结构,被GO紧密包裹.

图3 GO(a,b)和GO/PPy(c,d)的SEM(a,c)和TEM(b,d)图Fig.3 SEM(a,c)and TEM(b,d)images of GO(a,b)and GO/PPy(c,d)
分析GO/PPy插层复合物这种结构的形成原因和过程可能是:GO在超声波作用下先剥离成纳米片层,而MO与FeCl3在超声波作用下形成了纤维状络合物(MO-FeCl3),与GO层间的大量含氧官能团产生较强的相互作用力,有利于MO-FeCl3插入GO层间.接着Py单体在此纤维状络合物表面附着并以此为氧化剂和模板发生聚合反应,沿着模板生长,形成一维交错的PPy链状结构,同时,MO-FeCl3自降解,最终制得GO/PPy插层复合材料.整个过程示于图4.
3.4 电化学测试
电极材料的电化学性能不仅取决于材料本身的导电性、多孔性,还取决于电解液的性能.影响电解液选择的主要因素有:电阻、电容、稳定的电位窗口以及制造成本等.与有机电解液相比,水系电解液用于电容器安全性高、成本较低且无污染.本文选用1 mol·L-1Na2SO4和1 mol·L-1H2SO4溶液作为超级电容器的电解液不仅对环境友好、成本低,且与有机电解液相比具有较好的循环性能.
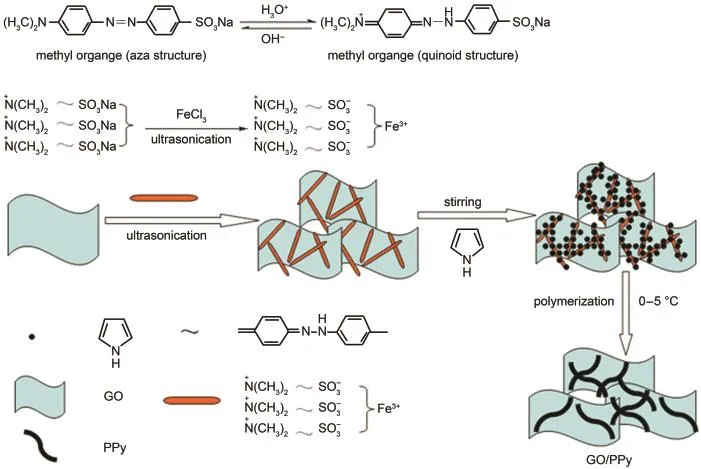
图4 GO/PPy插层复合材料形成过程示意图Fig.4 Schematic of the formation process of GO/PPy intercalation composite
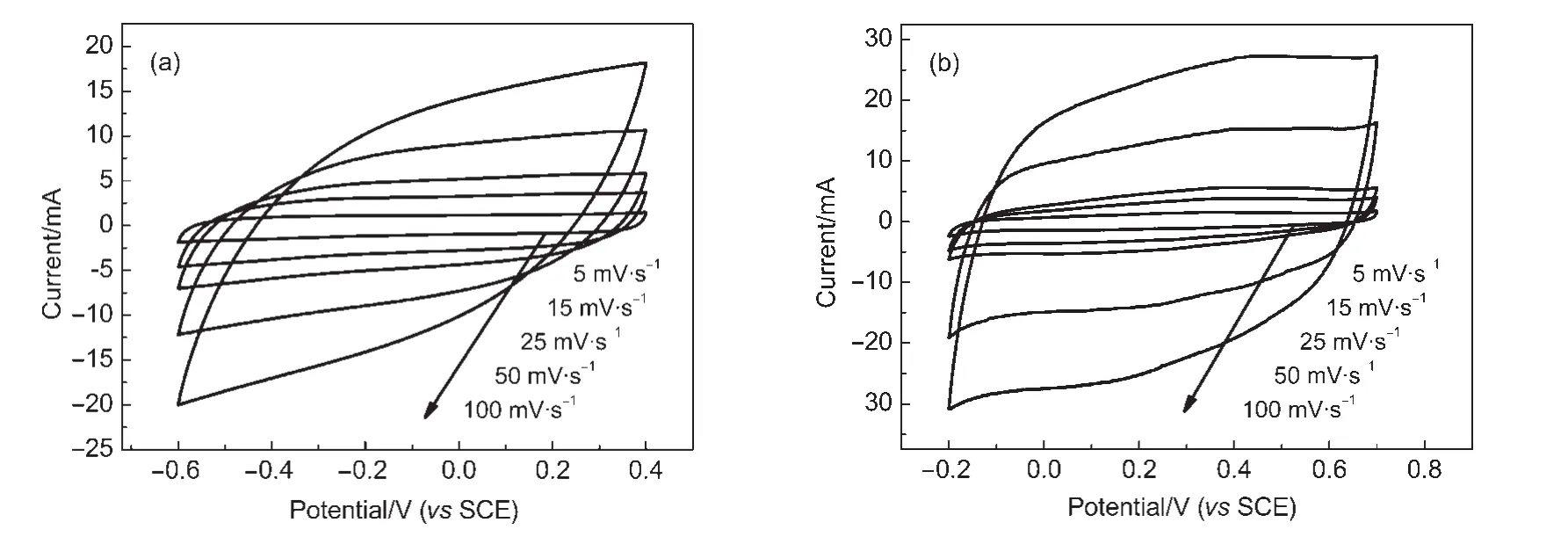
图5 GO/PPy复合材料在两种电解液中不同扫描速率时的循环伏安曲线图Fig.5 CV profiles of GO/PPy composite in two electrolytes at different scan rates(a)1 mol·L-1Na2SO4,(b)1 mol·L-1H2SO4
循环伏安分析是研究材料电化学特性的一个重要手段,图5为GO/PPy插层复合材料在1 mol·L-1Na2SO4和 1 mol·L-1H2SO4溶液中不同扫描速率下的循环伏安曲线图,电位区间分别为-0.6-0.4 V和-0.2-0.7 V.从图中可以看出,不同扫描速率下,GO/PPy复合材料在两种电解液中的循环伏安曲线都呈现出类似的“矩形”,具有对称性,说明该复合材料在两种电解液中均保持了良好的电化学特性且在电极表面有很好的电荷传递过程.但是,在1 mol·L-1Na2SO4电解液中,当扫描速率增大到100 mV·s-1时循环伏安曲线的“矩形”状略有变形,这是由于复合材料电极内部的阻抗使离子与电极间的相互作用减弱造成的,而这一影响在1 mol·L-1H2SO4电解液中要小得多.此外,相同扫描速率下,复合材料在1 mol·L-1H2SO4电解液中的循环伏安曲线的积分面积明显大于在1 mol·L-1Na2SO4电解液中的积分面积,表明复合材料在1 mol·L-1H2SO4电解液中有更大的电化学活性.
图6是电流密度为1 A·g-1时,纯GO、纯PPy和GO/PPy复合材料在两种不同电解液中的恒电流充放电曲线图,三者的质量比电容Cm可根据下列公式计算得到:
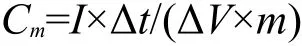
式中:Cm为复合电极材料的质量比电容(F·g-1);m是电极材料的质量(g);I为放电电流(A);Δt为放电时间(s);ΔV是电压区间(V).在1 mol·L-1Na2SO4电解液中GO、PPy和GO/PPy复合材料的比电容分别为14.8、249.6和310.9 F·g-1,而在1 mol·L-1H2SO4电解液中三者比电容分别为17.8、378.4和509.1 F·g-1.在两种不同电解液中,复合材料的比电容均远大于纯GO的比电容,也大于纯PPy的比电容.由于GO在复合物中添加的量很少,可见GO对复合材料总容量的贡献非常小,但其通过提高材料的导电性和分散性,增加了PPy的利用率从而提高材料的比容量和循环稳定性.
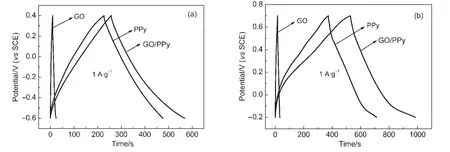
图6 纯GO、纯PPy和GO/PPy复合材料在两种电解液中的恒电流充放电曲线Fig.6 Galvanostatic charge/discharge curves of pure GO,pure PPy,and GO/PPy composite in two electrolytes
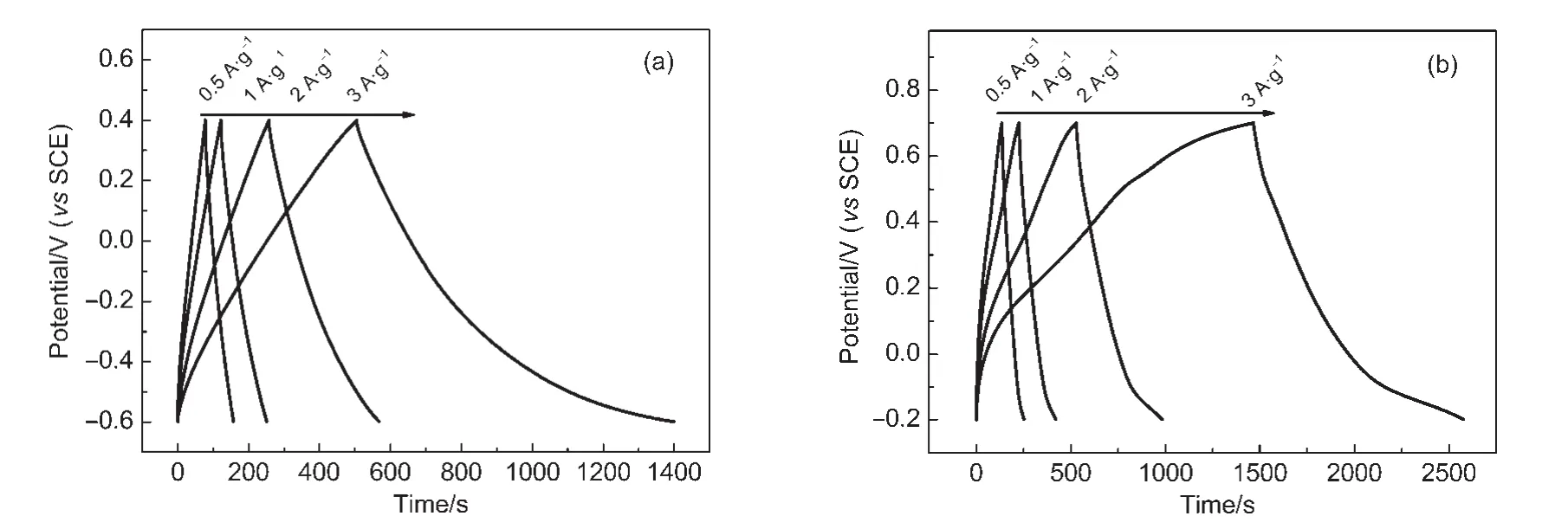
图7 GO/PPy复合材料在不同电流密度下的恒电流充放电曲线图Fig.7 Galvanostatic charge/discharge curves of GO/PPy composite at different current densities
图7是复合材料分别在两种电解液中不同电流密度下的首周恒电流充放电曲线图.由图可以看出,在整个充放电电位区间内,电位和时间都保持了很好的线性关系,图形基本对称,表明复合材料在两种电解液中的电化学反应具有良好的可逆性.当电流密度为0.5、1、2和3 A·g-1时,GO/PPy复合材料在1 mol·L-1Na2SO4电解液中的比电容分别为449.1、310.9、259.2和 238.8 F·g-1,在 1 mol·L-1H2SO4电解液中的比电容分别为619.0、509.1、435.0和392.3 F·g-1.硫酸电解液中的比电容稍大于硫酸钠电解液中的,与循环伏安测试结果相符.其原因为:H+离子在充放电过程中参与PPy掺杂与去掺杂的氧化还原反应,表现出更明显的赝电容行为,24,25如式(1)所示.
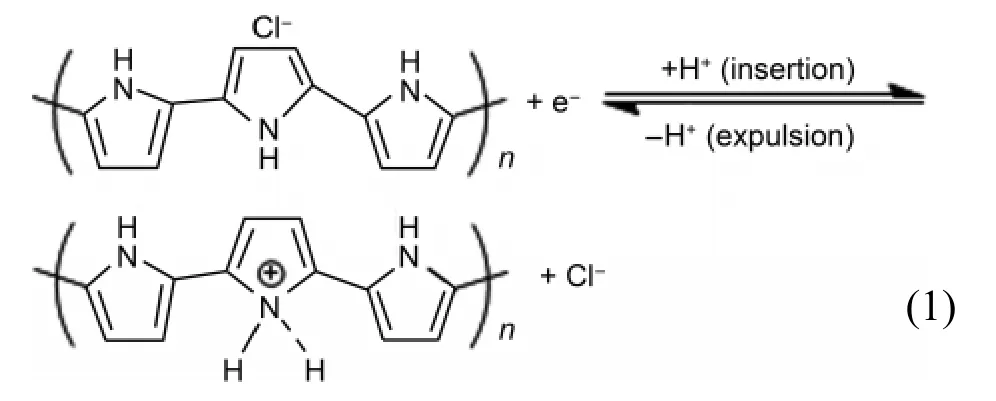
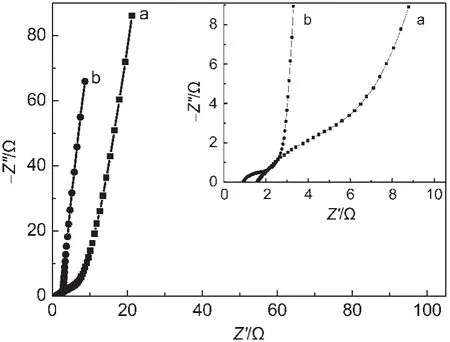
图8 GO/PPy复合材料在两种电解液中的交流阻抗图Fig.8 AC impedance plots of GO/PPy composite in two electrolytes
图8为GO/PPy插层复合材料在1 mol·L-1Na2SO4和1 mol·L-1H2SO4电解液中的交流阻抗图谱,测试的频率范围为yquist曲线由阻抗的实部(Z')和虚部(Z'')组成.在高频区,通过阻抗曲线与实轴的交点可以估算出复合材料电极的溶液电阻(Rs),半圆表示电荷在电极材料中传递过程,即电荷传递电阻(Rct),低频区45°的斜线部分是由电解液中的离子向电极表面扩散时所引起的Warburg阻抗.26,27由图8可知,GO/PPy复合材料在1 mol·L-1H2SO4和 1 mol·L-1Na2SO4电解液中的 Rs值分别约为1.0和1.6 Ω,表明复合材料在H2SO4溶液中的导电性和电容性能更好,与恒电流充放电测试结果相符.从右上角的内附图中可以看到,与Na2SO4电解液相比,复合材料电极在H2SO4电解液中的Warburg区域较小,说明在H2SO4电解液中,电极/电解液界面的电荷传递电阻小,电荷传递速率快.随着频率的降低,曲线b与实轴的夹角更接近90°,体现出复合材料电极在H2SO4电解液中的电化学行为更接近理想的电容行为.
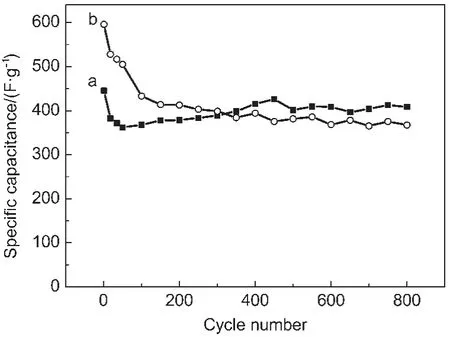
图9 GO/PPy复合材料在两种电解液中的循环性能图Fig.9 Cycle properties of GO/PPy composite in two electrolytes
图9为GO/PPy插层复合材料在1 mol·L-1Na2SO4和1 mol·L-1H2SO4电解液中,0.5 A·g-1电流密度下的循环寿命图.由图可知,虽然GO/PPy复合材料在1 mol·L-1H2SO4电解液中具有更好的电化学活性和更大的初始容量,但经过800次循环之后,复合材料电极的比电容保持了初始电容的62%,明显低于在Na2SO4电解液中达初始容量92%的容量保持率.与Na2SO4电解液相比,复合材料在H2SO4电解液中的比电容衰减较快,原因可能为:在酸性电解液中,PPy氧化(p-型)掺杂态的还原(脱掺杂)和再氧化(掺杂)伴随着H+的嵌入和脱出,此过程使GO/PPy插层结构不断膨胀和收缩,部分PPy可能从GO层间脱出,从而致使复合材料的容量损失较大.与文献12,28报道相比,复合材料在两种电解液中比电容的保持率均显著高于纯PPy,表明复合氧化石墨烯有效地改善了材料的循环稳定性.
4 结论
本文以FeCl3-MO为自降解模板,采用原位聚合法成功制备出GO/PPy插层复合材料.研究了复合材料在两种不同水系电解液(1 mol·L-1Na2SO4和1 mol·L-1H2SO4)中的电化学电容行为.电流密度为0.5 A·g-1时,复合材料在1 mol·L-1Na2SO4和1 mol·L-1H2SO4电解液中的比电容分别为449.1和619.0 F·g-1,经过800次循环稳定测试后,比容量保持率分别为92%和62%.测试结果表明,吡咯单体在GO层间交错聚合,阻碍了GO层与层间的堆叠,有利于充放电过程中离子和电子的传输,从而提高复合材料的比电容.同时,氧化石墨烯的加入能够缓解聚吡咯链在多次充放电过程中的膨胀和收缩,改善其循环稳定性.两者有效地发挥了各自优势及协同作用,展现出复合材料良好的电化学性能.
(1) Snook,G.A.;Kao,P.;Best,A.S.J.Power Sources 2011,196,1.doi:10.1016/j.jpowsour.2010.06.084
(2)Wang,G.P.;Zhang,L.;Zhang,J.J.Chem.Soc.Rev.2012,41,797.doi:10.1039/c1cs15060j
(3)Wang,J.P.;Xu,Y.L.;Zhu,J.B.;Ren,P.G.J.Power Sources 2012,208,138.doi:10.1016/j.jpowsour.2012.02.018
(4)Wang,J.P.;Xu,Y.L.;Wang,J.;Du,X.F.Synthetic Metals 2011,161,1141.doi:10.1016/j.synthmet.2011.01.011
(5)Zhang,D.C.;Zhang,X.;Chen,Y.;Yu,P.;Wang,C.H.;Ma,Y.W.J.Power Sources 2011,196,5990.doi:10.1016/j.jpowsour.2011.02.090
(6) Zhu,J.B.;Xu,Y.L.;Wang,J.;Wang,J.P.Acta Phys.-Chim.Sin.2012,28,373.[朱剑波,徐友龙,王 杰,王景平.物理化学学报,2012,28,373.]doi:10.3866/PKU.WHXB201112021
(7)Sahoo,S.;Nayak,G.C.;Das,C.K.Macromol.Symp.2012,315,177.doi:10.1002/masy.v315.1
(8) Li,L.Y.;Xia,K.Q.;Li,L.;Shang,S.M.;Guo,Q.Z.;Yan,G.P.J.Nanopart.Res.2012,14,908.doi:10.1007/s11051-012-0908-3
(9)Xu,J.J.;Wang,K.;Zu,S.Z.;Han,B.H.;Wei,Z.X.ACS Nano 2010,4,5019.doi:10.1021/nn1006539
(10) Xu,C.H.;Sun,J.;Gao,L.J.Mater.Chem.2011,21,11253.doi:10.1039/c1jm11275a
(11) Ding,B.;Lu,X.J.;Yuan,C.Z.;Yang,S.D.;Han,Y.Q.;Zhang,X.G.;Che,Q.Electrochimica Acta 2012,62,132.doi:10.1016/j.electacta.2011.12.011
(12) Zhang,H.Y.;Hu,Z.A.;Zhang,F.H.;Liang,P.J.;Zhang,Y.J.;Yang,Y.Y.;Zhang,Z.Y.;Wu,H.Y.Chinese Journal of Applied Chemisty 2012,29,674.[张海英,胡中爱,张富海,梁鹏举,张亚军,杨玉英,张子瑜,吴红英.应用化学,2012,29,674.]
(13)Zhu,C.Z.;Zhai,J.F.;Wen,D.;Dong,S.J.J.Mater.Chem.2012,22,6300.doi:10.1039/c2jm16699b
(14)Chang,H.H.;Chang,C.K.;Tsai,Y.C.;Liao,C.S.Carbon 2012,50,2331.doi:10.1016/j.carbon.2012.01.056
(15)Yang,X.M.;Zhu,Z.X.;Dai,T.Y.;Lu,Y.Macromol.Rapid Commun.2005,26,1736.
(16)Tang,L.H.;Wang,Y.;Li,Y.M.;Feng,H.B.;Lu,J.;Li,J.H.Adv.Funct.Mater.2009,19,2782.doi:10.1002/adfm.v19:17
(17)Feng,X.M.;Li,R.M.;Yan,Z.Z.;Liu,X.F.;Chen,R.F.;Ma,Y.W.;Li,X.A.;Fan,Q.L.;Huang,W.IEEE Transaction on Nanotechnology 2012,11,1080.doi:10.1109/TNANO.2012.2200259
(18) Fan,Z.J.;Kai,W.;Yan,J.;Wei,T.;Zhi,L.J.;Feng,J.;Ren,Y.M.;Song,L.P.;Wei,F.ACS Nano 2011,5,191.doi:10.1021/nn102339t
(19)Zhang,L.L.;Zhao,S.Y.;Tian,X.N.;Zhao,X.S.Langmuir 2010,26,17624.doi:10.1021/la103413s
(20)Gu,Z.M.;Li,C.Z.;Wang,G.C.;Zhang,L.;Li,X.H.;Wang,W.D.;Jin,S.L.Journal of Polymer Science Part B:Polymer Physics 2010,48,1329.doi:10.1002/polb.v48:12
(21)Hummers,W.S.;Offeman,R.E.J.Am.Chem.Soc.1958,80,1339.doi:10.1021/ja01539a017
(22) Zu,S.Z.;Han,B.H.J.Phys.Chem.C 2009,113,13651.
(23) Meyer,J.C.;Geim,A.K.;Katsnelson,M.I.;Novoselov,K.S.;Obergfell,D.;Roth,S.;Girit,C.;Zettl,A.Solid State Communication 2007,143,101.doi:10.1016/j.ssc.2007.02.047
(24) Tian,Y.;Yang,F.L.;Yang,W.S.Synthetic Metals 2006,156,1052.doi:10.1016/j.synthmet.2006.06.023
(25) Cai,Y.M.;Qin,Z.Y.;Chen,L.Progress in Natural Science:Material International 2011,21,460.doi:10.1016/S1002-0071(12)60083-5
(26) Sahoo,S.;Karthikeyan,G.;Nayak,G.C.;Das,C.K.Synthetic Metals 2011,161,1723.
(27)Zhang,K.;Zhang,L.L.;Zhao,X.S.;Wu,J.S.Chem.Mater.2010,22,1392.doi:10.1021/cm902876u
(28) Sahoo,S.;Dhibar,S.;Das,C.K.Express Polymer Letters 2012,6,965.doi:10.3144/expresspolymlett.2012.102
