3,4-吡啶二酸钡配合物的合成、结构、荧光和热稳定性研究
2013-09-15冯建华李会会
左 锣 冯建华 郭 莉 李会会 吴 刚*,,2
(1滁州学院材料与化学工程学院,滁州 239012)
(2南京大学配位化学国家重点实验室,南京 210093)
The rational design and syntheses of metalorganic frameworks have been of increasing interest in the crystal engineering of coordination polymers owing to their ability to provide with fascinating topological structures and material properties,which may render them for potential applications in non-linear optical,magnetic,and luminescent materials[1-4].However,to predict and further control the framework array of a given crystalline product still remain a considerable challenge at this stage.This mainly arises from the fact that the subtle assembled progress may be influenced by many intrinsic and external parameters,such as solvent[5-6],concentration[7],counteranion[8],temperature[9],pH value of the solution[10],and so on.However,it is very significant to select befitting ligands and metal ions in the assembly of coordination polymers. Carboxylate-containing ligands have particularly aroused much more attention because of diverse coordination modes of carboxylate groups to the metal ions,which can adopt a variety of bonding modes to metals,including (i)terminal monodentate,(ii)chelating to a single metal,(iii)bridging bidentate(iv)bridging tridentate and so on.Generally,multicarboxyl acids can be utilized in the formation of different dimensional coordination polymers[11-14].
On the other hand,most of the research works so far are mainly focused on the incorporation of transition metal ions and rare-earth ions as coordination centers.Relatively less attention has been paid to the alkaline earth metal-based compounds[15].However,compared with divalent transition metal ions,alkaline earth metal ions,for example,strontium(Ⅱ)and barium(Ⅱ),have larger ionic radius and therefore could adopt a higher coordination number, leading to an interesting topological arrangement.In addition,the main group alkaline-earth metals,owing to their low polarizability and various coordination models for the construction of polymer materials featuring robust structures and interesting properties,are receiving more and more research attention recently[16-17].In this manuscript,wedescribesyntheses,structure,properties of an alkaline earth metal barium coordination polymer[Ba2(pdc)2(H2O)3]n(1)of 3,4-pyridinedicarboxylic acid(H2pdc).
1 Experimental
1.1 Reagents and physical measurements
All reagents commercially available were of reagent grade and used without further purification.Solvents were purified according to the standard methods.C,H and N element analyses were carried out on a Perkin-Elmer 240C elemental analyzer.IR spectra were recorded on a Nicolet 6700 FT-IR spectrophotometer by using KBr pellet in the range of 4 000~400 cm-1.Thermogravimetric analyses(TGA)were carried out with a SDT Q600 instrument under 100.0 mL·min-1flowing nitrogen,and ramp rate of 20.00 ℃·min-1from 26 to 900 ℃.The luminescent spectra of the solid samples were recorded at room temperature on a CaryEclipse 300 spectrophotometer with a xenon arc lamp as the light source.In the measurements of the emission and excitation spectra,the passage width is 10.0 nm.Powder X-ray diffraction patterns were performed with a Bruker D8 ADVANCE X-ray diffractometer with Cu Kα1radiation at 40 kV and 40 mA.
1.2 Synthesis of the title complex[Ba2(pdc)2(H 2O)3]n(1)
The mixture of 3,4-pyridinedicarboxylic acid 16.7 mg(0.1 mmol)and BaCl2·2H2O 24.4 mg(0.1 mmol)in mixture solution of 3 mL distilled H2O and 5 mL DMF(N,N,-dimethylformamide)was stirred about 30 min at room temperature.Then the mixture was sealed in a 25 mL Teflon-lined stainless steel container and heated at 160 ℃ for 3 d.The colorless block crystals were obtained (15.1 mg).Anal.Calcd.for C14H12Ba2N2O11(658.94)(%):C,25.52;H,1.84;N,4.25.Found(%):C,25.49;H,1.88;N,4.26.FT-IR spectrum was recored as KBr pellets on a 6700 FT-IR spectrophotometer(KBr pellet,cm-1):3 410(bs),1 608(s),1 566(s),1 524(w),1 459(s),1 443(m),1 162(m),1 066(w),888(m),850(m),793(m),726(w).
1.3 Crystal structure determination
The crystallographic data collections for complex 1 with dimensions of 0.34 mm ×0.29 mm ×0.24 mm were carried out on a Bruker Smart ApexⅡCCD with graphitemonochromated Mo Kα radiation(λ=0.071 073 nm)at 296(2)K using the ω-scan technique.The structure was solved by direct methods using the SHELXS-97 program;and all non-hydrogen atoms were refined anisotropically on F2by the full-matrix leastsquares technique using the SHELXL-97 crystallographic software package[18-19].The hydrogen atoms were generated geometrically.The details of the crystal parameters,data collection and refinement for this compound are summarized in Table 1.Selected bond lengths and bong angles for complex 1 are listed in Table 2.
CCDC:887479.
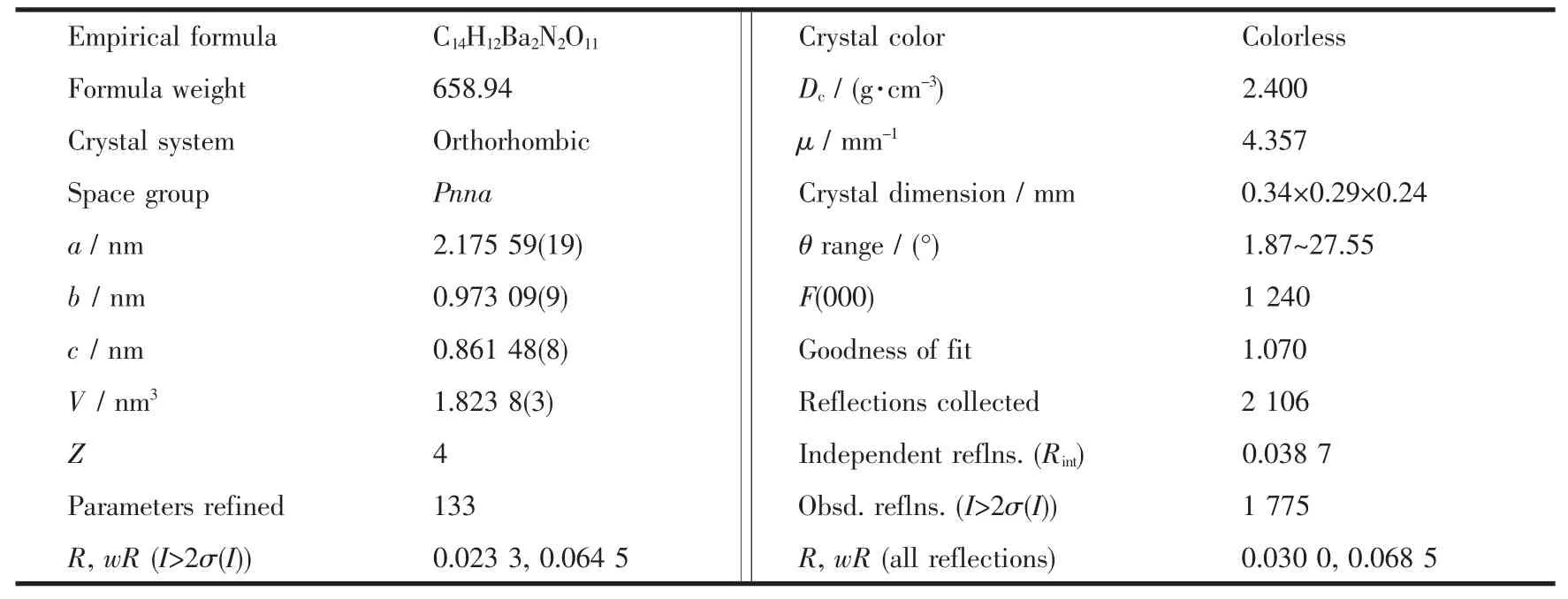
Table 1 Crystallographic data for complex 1
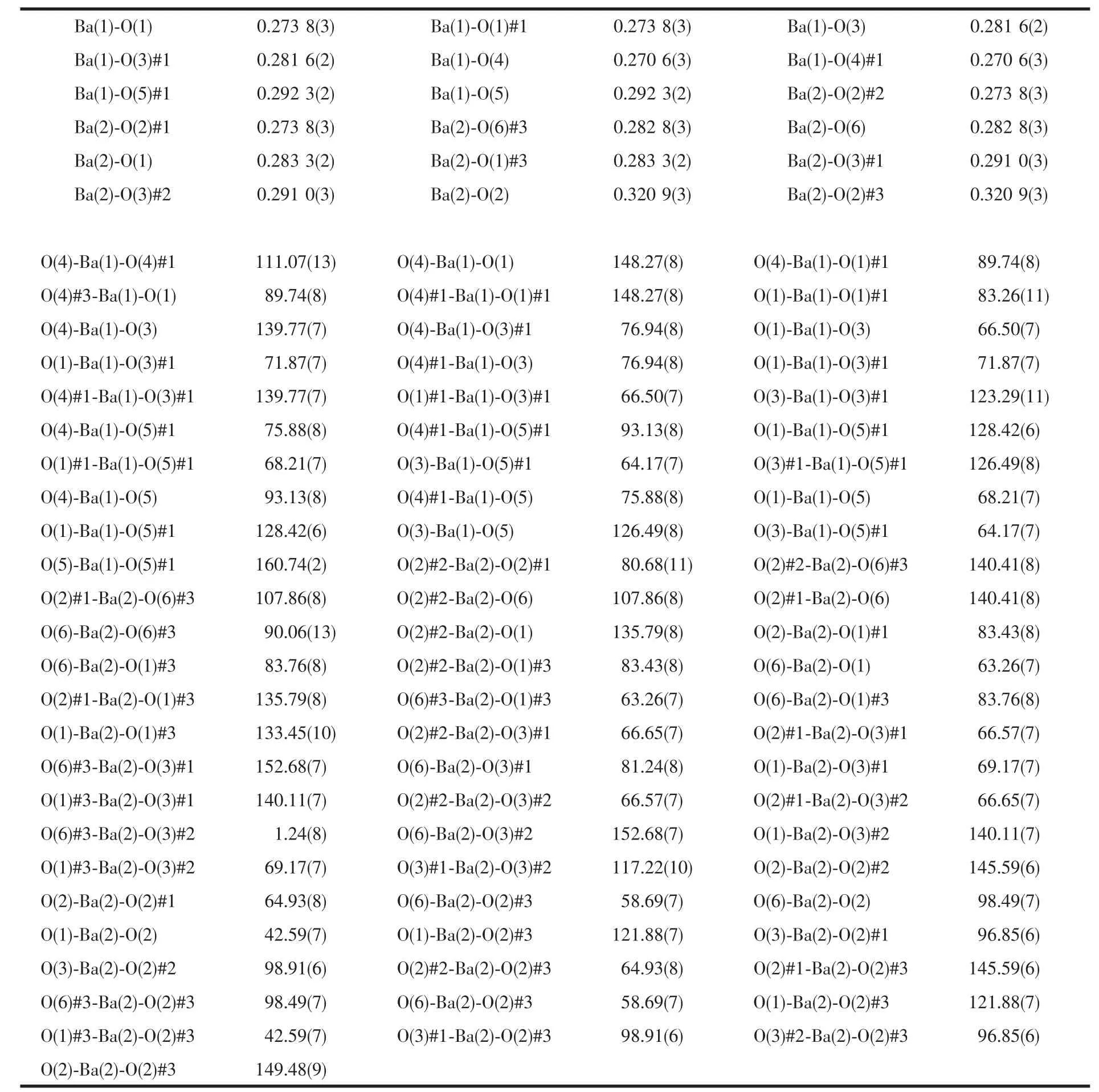
Table 2 Selected bond lengths(nm)and bond angles(°)for complex 1
2 Results and discussion
2.1 IR spectra
The strong and broad absorption bands in the range of 3 410 cm-1for 1 is assigned as the characteristic peaks of OH vibration.The strong vibrations around 1 608 and 1 566 cm-1for 1 correspond to the asymmetric and symmetric stretching vibrations of carboxylate group,respectively.The absence of strong bands ranging from 1 690 to 1 730 cm-1indicates the deprotonation of ligand H2pdc,which agrees with the structural results.
2.2 Crystal structure of 1
Titled compound 1 crystallized in theorthorhombic space group Pnna,and has the 2D network structure.In complex 1,there are two Ba(Ⅱ)ions,two pdc2-anion and three coordinated water molecules per formula unit.Because the site occupation factor of Ba1,Ba2 is 0.5,respectively,asymmetric unit only contains one Ba(Ⅱ) atom and a pdc2-anion.The Ba1 center is coordinated by eight oxygen atoms of six carboxylate groups from four pdc3-anions and twoμ2-bridging coordinated water molecules to form a distorted square anti-prism (Fig.1).The distortion of the structure is clearly evident from the different Ba1-O bond lengths and O-Ba1-O bond angles,which can be divided into six shorter(0.270 6(3)~0.281 6(2)nm)and two longer bonds(0.292 3(2)nm),and the O-Ba1-O bond angles around the metal center range from 64.17(7)°to 148.27(8)°.
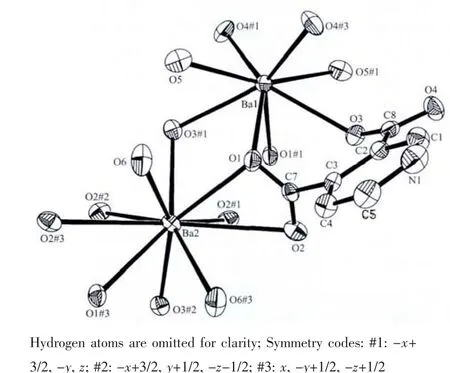
Fig.1 ORTEPview of coordination environment of Ba(Ⅱ)atom in 1 with 50%probability displacement
Each Ba2 atom is 10-coordinate and in a distorted dicapped square prism coordination geometry,coordinating to eight carboxyl O atoms from six different carboxyl groups of four different pdc2-anions,two oxygen atoms from two coordinated molecules (Fig.1).The Ba2-O bond distances are in the range of 0.273 8(3)~0.320 9(3)nm,and the OBa2-O bond angles around the metal center vary from 42.59(7)°to 152.68(7)°.The distortion of the structure is also clearly evident from the different Ba2-O bond lengths,which can be divided into six shorter(0.273 8(3)~0.283 3(2)nm)and four longer bonds(0.291 0(3)~0.320 9(3)nm),The Ba-O distances in compound 1 are of the same order of magnitude as in comparable complexes[20-22].The N atom of pyridine does not coordinate with Ba2+ion,which takes part in forming O-H…N hydrogen bond,resulting in 3D structure.
In ligand pdc2-anion,two carboxylate groups of pdc2-are not coplanar with the central pyridine ring.The angles of pyridine ring plane between two carboxylate groups are 87.8°,8.8°,respectively.Two carboxylate groups of pdc2-adopt two different coordination modes.One takes μ3-η2∶η1bridging coordination mode,while the other takes μ3-η2∶η2bridging coordination mode.The whole pdc2-anion and μ2-bridged water molecules connect Ba(Ⅱ)atoms,giving rise to a 2D network structure (Fig.2).Then 2D layers further are linked together to give rise to 3D structure by O-H…N hydrogen bonds(Fig.3,Table 3).
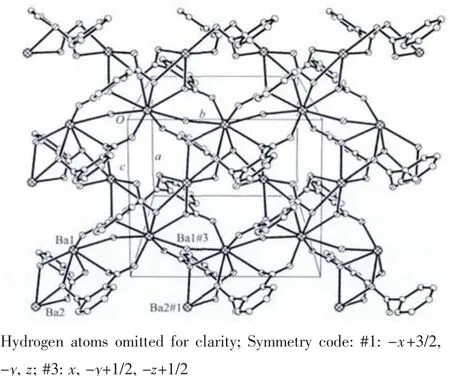
Fig.2 Two-dimensional layer of 1
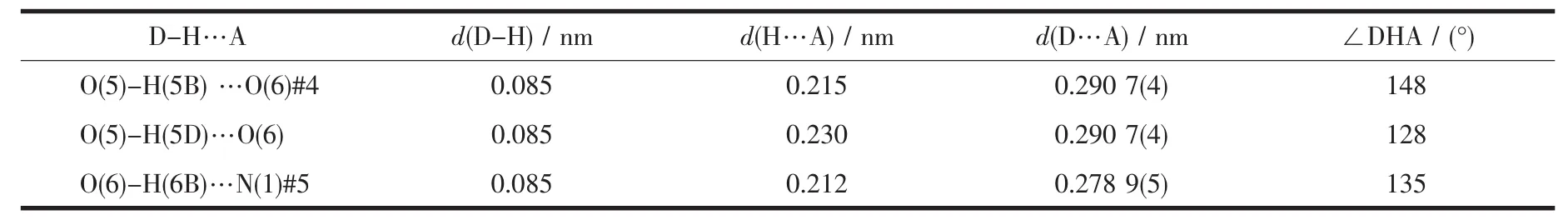
Table 3 Distances and angles of hydrogen bonds for complex of 1
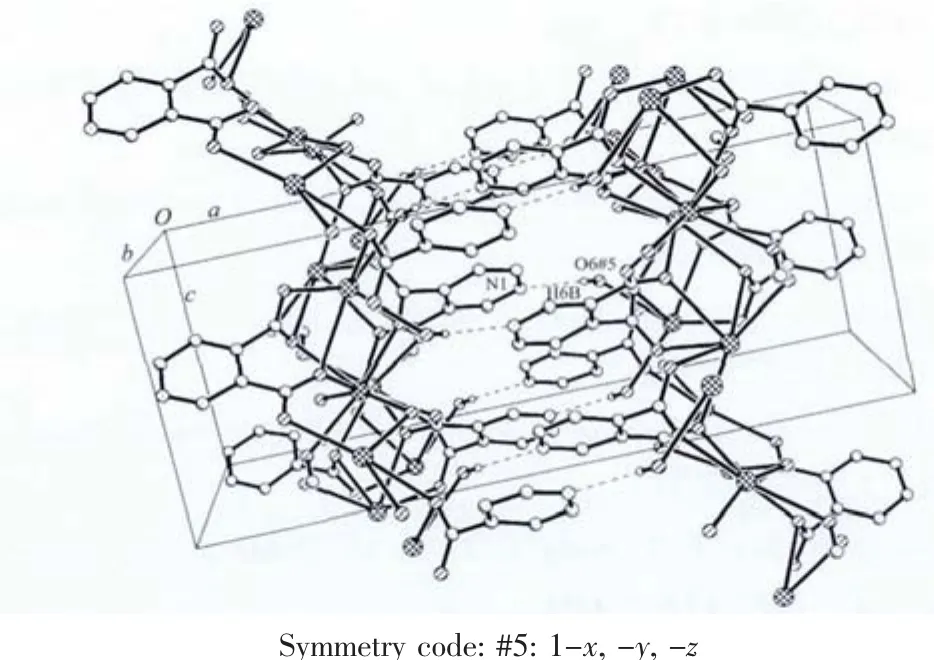
Fig.3 Three-dimensional structure of 1 stabilized by O-H…N hydrogen bonding interactions
3.1 Luminescent property of complex 1
The fluorescent properties of the H2pdc ligand and its Ba(Ⅱ)complex 1 were studied in the solid state at room temperature (Fig.4).The maximum of the emission for the complex 1 is located at 450 nm when exited with the light of 300 nm.To ascertain the adscription of emission spectra,the photoluminescence of pure H2pdc was measured under the same conditions.However,the free H2pdc ligand almost does not display emission at this condition.Such fluorescent behavior of complex 1 suggests that the emission may be mainly attributed to π-π*intraligand fluorescence.
3.2 Thermal analysis
Typical curve for 1 is presented in Fig.5.In the temperature range 26~218 ℃,there is only one weight-loss of 7.73%with an endothermal peak that corresponds to the successive release of three coordinated water per formula,which is in good agreement with the theoretical weight-loss value(7.73%).The anhydrous product is stable between 218~477 ℃.However,there is a weight-loss of 23.43% (theoretical,24.01%)observed from 477 to 716℃attributed to the release of the two pyridine molecules,resulting from decomposition of ligand.In this step,BaCO3is formed.Finally,there is exothermic peak at 733℃,which is ascribed to the decomposition of the BaCO3to the metal oxide BaO.The TGcurve of 1 is consistent with the formula,[Ba2(pdc)2(H2O)3]n,and in agreement with the single crystal X-ray analysis.
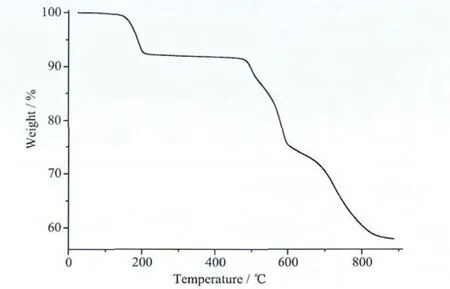
Fig.5 TG curve of 1
Furthermore, dehydration and rehydration experiments were performed for 1 and powder X-ray diffraction(PXRD)was used to check the phases(Fig.6).1 was heated at about 218 ℃ for 1 h,to obtain complete loss of water.Then,the dehydrated phase was afterwards exposed to water vapor at room temperature for another 24 h.From the Fig.6,the powdered dehydrated phase has a PXRD pattern different from that of complex 1 (Fig.6),indicating that the framework of 1 collapses after removal of coordinated water.Also from the Fig.6,we can see the PXRD pattern of rehydrated sample is different from the diffraction pattern of 1,which shows that the dehydration and rehydration processes for 1 are irreversible.
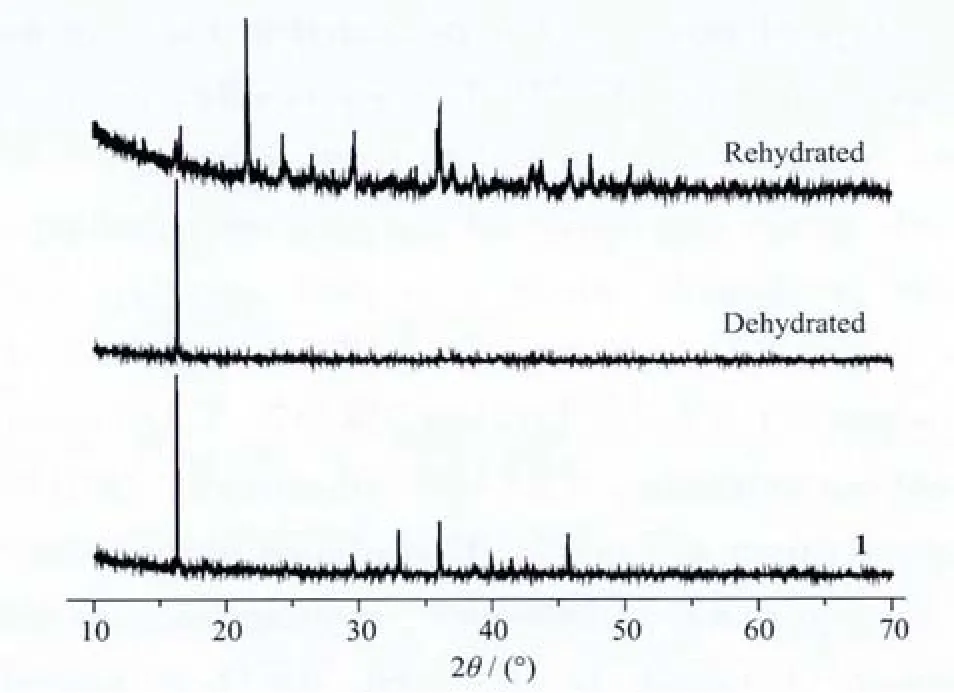
Fig.6 Powder X-ray diffraction patterns of 1,the dehydrated and the rehydrated
[1]LI Jing(李静),JI Chang-Chun(季长春),WANG Zuo-Wei(王作为),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2009,25:2083-2089
[2]ZHU Ying-Gui(朱英贵),JU Xue-Hai(居学海),SONG Ying-Lin(宋瑛林),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2008,24(12):2029-2034
[3]Kong CY.J.Inorg.Organomet.Polym.,2011,21:189-194
[4]Niu C Y,Pan Z L,Dang Y L,et al.J.Inorg.Organomet.Polym.,2011,21:611-618
[5]Baxter PN W,Khoury R G,Lehn JM,et al.Chem.Eur.J.,2000,6:4140-4148
[6]Long D L,Blake A J,Champness N R,et al,Chem.Eur.J.,2002,8:2026-2033
[7]Su CY,Cai Y P,Chen CL,et al.J.Am.Chem.Soc.,2003,125:8595-8613
[8]Ciurtin D M,Pschirer N G,Smith M D,et al.Chem.Mater.,2001,13:2743-2745
[9]Baxter P N W,Lehn J M,Baum G,et al.Chem.Eur.J.,2000,6:4510-4517
[10]Pan L,Huang X Y,Li J,et al.Angew.Chem.,Int.Ed.,2000,39:527-530
[11]Abuhmaiera R,Lan Y,Ako A M,et al.CrystEngComm,2009,11:1089-1096
[12]Motreff A,da Costa R C,Allouchi H,et al.Inorg.Chem.,2009,48:5623-5625
[13]Berggren G,Anderlund M F,Styring S,et al.Inorg.Chem.,2012,51:2332-2337
[14]Bo Q B,Sun Z X,Song G L,et al.J.Inorg.Organomet.Polym.,2007,17:615-622
[15]Wang F,Liu Z S,Yang H,et al.Angew.Chem.,Int.Ed.,2011,50:450-453
[16]Cao R,Lu J,Batten SR,CrystEngComm,2008,10:784-789
[17]Zhang X,Huang Y Y,Zhang M J,et al.Cryst.Growth Des.,2012,12:3231-3238
[18]Sheldrick G M.SHELXS 97,Program for the Solution of Crystal Structure,University of Göttingen,Germany,1997.
[19]Sheldrick G M.SHELXL 97,Program for the Refinement of Crystal Structure,University of Göttingen,Germany,1997.
[20]Fromma K M,Goesmann H.Acta Cryst.C56,2000:1179-1180
[21]Yu JO,CtéA P,Enright G D,et al.Inorg.Chem.,2001,40:582-583
[22]Zhu H F,Zhang Z H,Sun W Y,et al.Cryst.Growth Des.,2005,5:177-182