集胞藻PCC6803基因slr2049定点突变及其体内重组功能研究
2013-08-29陈思礼
朱 超 陈思礼
(中南民族大学 生命科学学院,武汉 430074)
藻胆蛋白是蓝藻中一种捕光色素蛋白,由藻胆色素(Phycobilin)与其相应的脱辅基蛋白中保守性半胱氨酸的巯基以硫醚键共价结合而成.藻蓝蛋白只有在结合了色基的情况下才有光学活性,而色基连接到脱辅基蛋白半胱氨基酸上需要色素裂合酶的催化[1-3].定点突变能够通过改变特定的氨基酸获得突变蛋白质,研究蛋白质的结构与功能,从微观水平上阐明正常状态下基因的调控机理、疾病的病因和机理[4].可以利用定点突变对合成藻胆蛋白基因进行改造,以便深入了解光合作用中各种藻胆蛋白捕获和传递光能的精细机制[5].藻蓝蛋白不仅可作为荧光探针用于免疫检测、荧光显微术和流式细胞仪技术,作为光敏剂用于光动力治疗癌症等方面,还具有提高机体免疫力、抗氧化,防治肿瘤等多种生理活性[6-9].
通过BLAST 软件同源性分析,从集胞藻PCC 6803中发现与裂合酶编码基因cpeS具有同源性的基因slr2049.本实验利用定点突变技术构建slr2049的8个突变体,以研究这8个氨基酸位点对酶催化活性的影响,确定哪些氨基酸在slr2049 酶催化活性中起到关键作用,从而为slr2049结构和功能的研究提供相关信息.
1 材料和方法
1.1 材料与试剂
集胞藻PCC6803购自中国科学院水生所藻种库,大肠杆菌E.coli,DH5α,BL21(DE3)菌株均为本实验室保存,克隆载体pBluescipt II SK(+),表达载体pCDFDuet-1 为本实验室保存,表达载体pET-23a(+)由华中师范大学生科院惠赠.限制性内切酶Pst I、Sal I、EcoR I和Xho I,T4DNA 连接酶,DL2000,DL5000,TaKaRa MutanBEST Kit定点突变试剂盒均为TaKaRa产品.DL250 购于武汉力博瑞生物科技有限公司.DNA 回收试剂盒,质粒小量快速提取试剂盒均为Axygen 公司产品.Taq酶、蛋白Marker为Fermentas公司产品.亲和层析介质购自南京金斯瑞生物科技有限公司.
1.2 实验方法
1.2.1slr2049、ho1、cpcB、pcyA4 个基因的PCR扩增 根据NCBI提供的基因序列,利用Primer premier 5软件设计引物,并在上下游引物分别引入酶切位点(下划线部分),分别由南京金斯瑞生物科技有限公司和擎科新业生物技术有限公司合成.
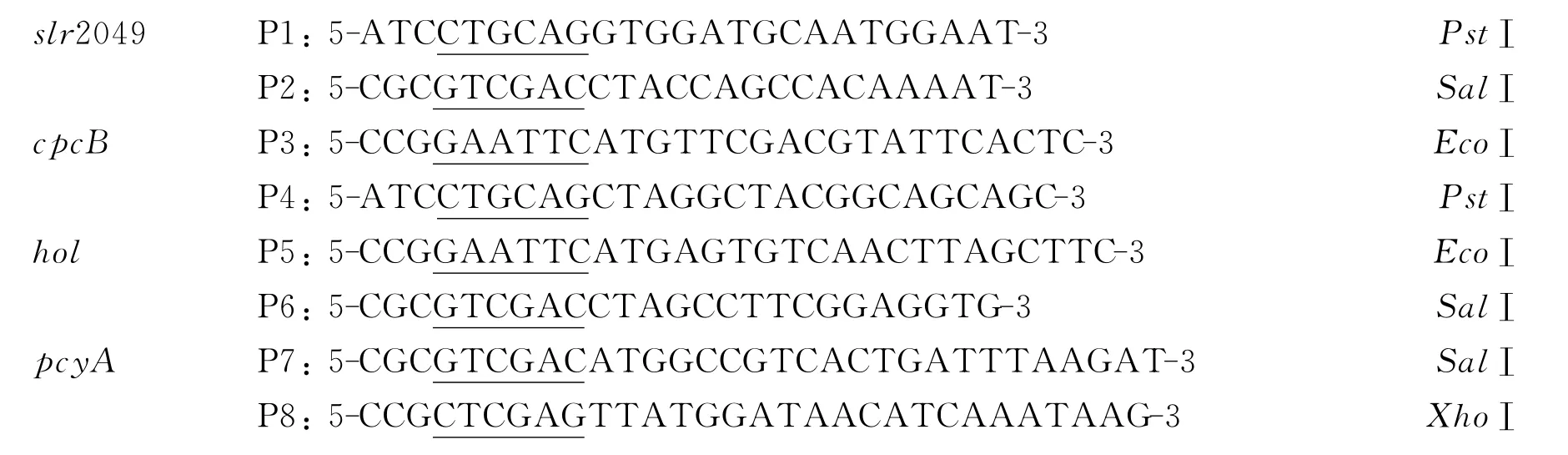
提取的集胞藻PCC6803 总DNA 为模板,利用上述引物进行Touch-down PCR 扩增4 个基因.slr2049、ho1 和cpcB的PCR扩增反应程序:94℃5 min;94℃1 min,55℃~52℃(每循环降0.5℃)1min,72℃2min循环10次;94℃1min,52℃1min,72℃2min循环20次;72℃10min.pcyA 的PCR 扩增程序:94℃5 min;94℃1 min,60℃~50℃(每循环降0.5℃)1 min,72℃2 min循环20次;94℃1min,50℃1min,72℃2min循环15次;72℃10min.
1.2.2 4个基因的克隆 4个PCR 目的产物经1%琼脂糖凝胶电泳回收,连接pBluescript II SK(+)载体,转化感受态大肠杆菌DH5α,氨苄青霉素抗性筛选阳性克隆,挑取菌落培养于含氨苄青霉素的LB培养基中,经过菌落PCR、双酶切重组质粒鉴定,送南京金斯瑞测序验证.
1.2.3slr2049突变体的构建 将slr2049 的蛋白序列与已经报道的6种色素裂合酶蛋白序列进行比对,得到22 个相同的位点.以构建的克隆载体pBlue-slr2049野生型为模板,设计引物(下划线是突变位点),使用定点突变试剂盒对8个位点进行点突变.操作过程参照试剂盒使用说明书.引物由擎科新业生物技术有限公司合成,测序验证.

1.2.4 表达载体pCDF-cpcB-slr2049野生型和突变体以及pET-ho1-pcyA的构建、蛋白的表达及条件优化EcoRⅠ与PstⅠ分别双酶切克隆载体pBlue-cpcB和pCDFDuet-1原核表达载体,EcoRⅠ与SalⅠ分别双酶切克隆载体pBlue-ho1 和pET-23a(+)原核表达载体,回收片段、连接、转化至BL21(DE3)感受态细胞,分别用链霉素及氨苄青霉素抗性筛选,测序验证.
PstⅠ和SalⅠ分别双酶切克隆载体pBlueslr2049野生型、pBlue-slr2049突变体和重组子pCDF-cpcB,SalⅠ和XhoⅠ分别双酶切克隆载体pBlue-pcyA和重组子pET-ho1,重复上述过程.
培养过夜的重组质粒pCDF-cpcB-slr2049野生型和突变体以及pET-ho1-pcyA分别以1∶50接种于含相应抗生素的LB培养基中,37℃继续振荡培养至OD600=0.4~0.6,加IPTG(终浓度分别为(0.6mmol/L和0.8mmol/L)、28℃诱导表达10h;同时用未加IPTG的重组菌和含pCDFDuet-1及pET-23a(+)空载体的BL21(DE3)菌为对照.离心收集细胞,含重组子的细胞重悬于超声波缓冲液中(pH7.0),超声6min,离心得到上清和沉淀,ddH2O稀 释,加 上样缓冲液、β-巯基乙醇煮沸6min,进行SDS-PAGE检测.
1.2.5 重组藻蓝蛋白β亚基的体内合成及蛋白诱导 pCDF-cpcB-slr2049野生型和突变体分别和pET-ho1-pcyA共转化入BL21(DE3)的感受态细胞中,含有链霉素和氨苄青霉素的双抗平板进行筛选,挑取菌落培养于含链霉素和氨苄青霉素的LB培养基中,37℃振荡培养过夜.
将培养过夜的共转化重组菌以1∶50接种于含相应抗生素的LB培养基中,37℃继续振荡培养至OD600=0.4~0.6,加IPTG(终浓度为1mmol/L)、20℃诱导表达12h;同时用未加IPTG的共转化重组菌和含pCDFDute-1及pET-23a(+)空载体的BL21(DE3)菌为对照.处理同上,进行SDS-PAGE检测重组蛋白.
1.2.6 重组藻蓝蛋白β亚基表达和纯化 培养过夜的共转化重组菌以1∶20接种于含双抗的1.1L LB 培养基中,37℃继续振荡培养至OD600=0.4~0.6,加IPTG(终浓度为1mmol/L)、20℃诱导表达12h;离心收集细胞,细胞重悬于LE buffer(50mmol NaH2PO4,300mmol NaCl,pH 8.0),超声破碎40min,离心得到上清,参照亲和层析说明书操作,得到的蛋白样品用SDS-PAGE检测.
1.2.7 光谱测定 吸收光谱用UW757CRT 型紫外可见分光光度计,紫外可见光谱扫描范围300~800nm,扫描速度960nm/min,狭缝宽度1.0nm;荧光光谱用F-2500荧光光度计,荧光光谱扫描速度1500nm/min,狭缝宽度5.0nm.
2 实验结果
2.1 slr2049、ho1、cpcB、pcyA4个基因的克隆与鉴定
以集胞藻Synechocystissp.PCC 6803基因组DNA 为模板,P1-P8引物分别扩增出基因slr2049、cpcB、ho1、pcyA,经1%的琼脂糖凝胶电泳鉴定,得到4个条带和NCBI上已知基因大小分别是579bp,519bp,723bp,747bp相符.回收扩增的产物,连接、转化得到重组质粒,采用菌落PCR 及限制性内切酶双酶切作进一步鉴定(图1),通过测序证明得到了目的条带.

图1 4个基因的PCR 产物,pBlue-slr2049野生型、pBlue-cpcB、pBlue-pcyA、pBlue-ho1 双酶切鉴定图谱Fig.1 PCR products of the four genes and Enzymatic cleavage analysis of pBlue-slr2049、pBlue-cpcB、pBlue-pcyA、pBlue-ho1
2.2 slr2049突变体鉴定
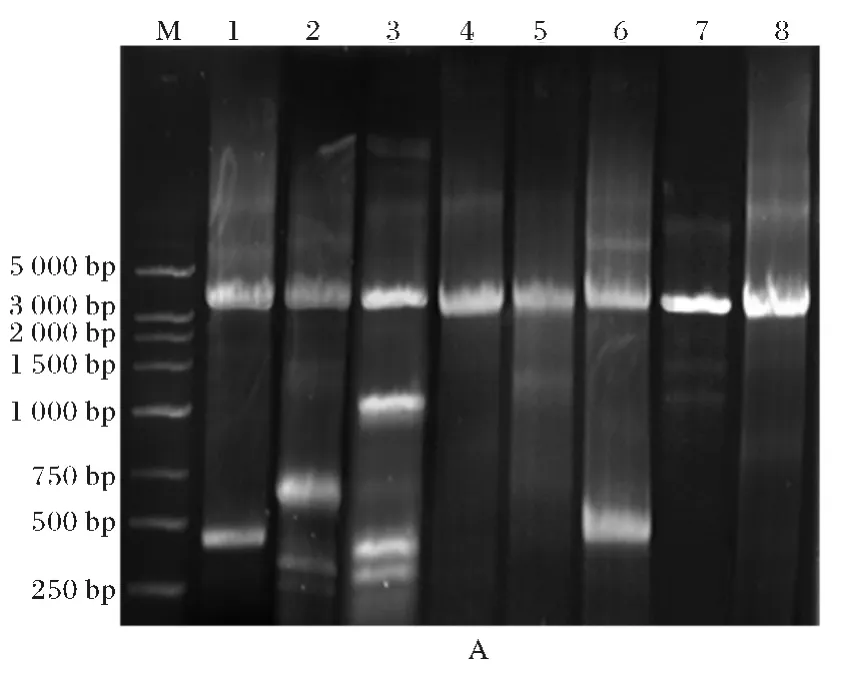
以pBlue-slr2049野生型为模板,采用定点突变引物P9-P24扩增突变基因,挑取阳性克隆,进行菌落PCR,PstⅠ、SalⅠ双酶切和测序鉴定,证明突变体构建成功(图2).
2.3 重组表达载体pCDF-cpcB-slr2049野生型和突变体以及pET-ho1-pcyA 构建与表达
重组质粒pBlue-cpcB、pBlue-ho1经双酶切并回收基因后亚克隆到原核表达载体pCDFDuet-1、pET-23a(+),构建重组表达质粒pCDF-cpcB、pET-ho1.克隆质粒pBlue-slr2049 野生型、pBlueslr2049突变体以及pBlue-pcyA 经相应的限制性内切酶双酶切,重复上述操作.成功构建重组表达载 体pCDF-cpcB-slr2049野生型和突变体以及pET-ho1-pcyA.
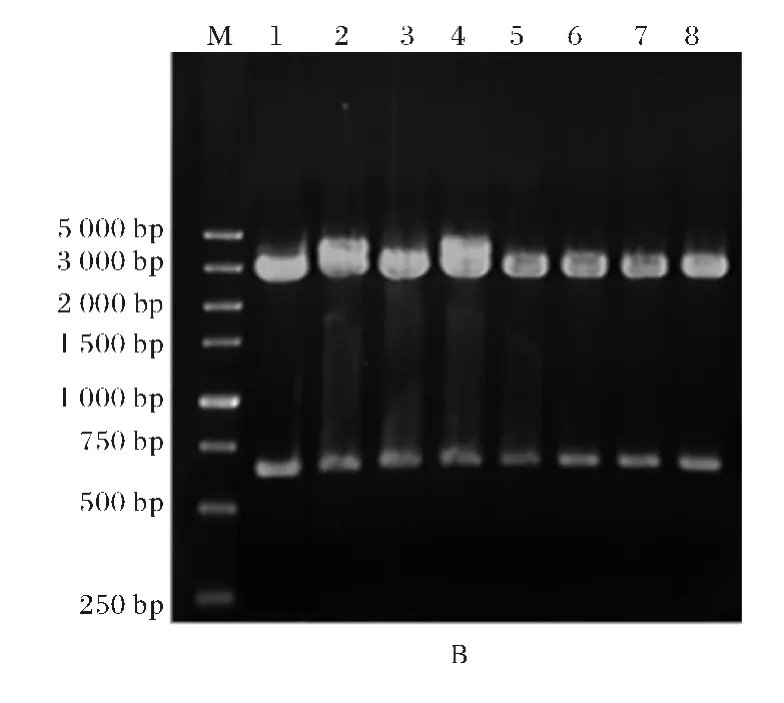
图2 8个slr2049突变体PCR 和pBlue-slr2049突变体双酶切鉴定图谱Fig.2 PCR products of the eight slr2049mutants and Enzymatic cleavage analysis of pBlue-slr2049mutants
图3 表达载体双酶切鉴定图谱Fig.3 Enzymatic cleavage analysis of expression vector
重组表达质粒pCDF-cpcB-slr2049 野 生 型 和突变体以及pET-ho1-pcyA 转化E.coli后,筛选阳性重组子,经IPTG 诱导表达.SDS-PAGE 分析表达产物(图4),可以看出,目的蛋白的大小与预期的蛋白分子量吻合.CpcB、Slr2049野生型、Slr2049突变体和PCB合成酶表达成功.

图4 pCDF-cpcB-slr2049野生型和pET-ho1-pcyA 表达产物的SDS-PAGE电泳图Fig.4 Analysis of expressed products of pCDF-cpcBslr2049and pET-ho1-pcyA SDS-PAGE
2.4 重组藻蓝蛋白β亚基的体内合成及蛋白诱导、纯化和光谱分析
将重组质粒pCDF-cpcB-slr2049野生型和突变体分别与pET-ho1-pcyA共转化到大肠杆菌BL21(DE3)后,筛选阳性重组子,经IPTG 诱导表达,得到共转化菌株,冷冻离心收集菌体后,发现含有野 生型重组质粒pCDF-cpcB-slr2049 和 突 变 体重组质粒pCDF-cpcB-slr2049(H21S)/slr2049(L23S)/slr2049(F25S)/slr2049(W72L)/slr2049(G84S)/slr2049(Y124)的菌体沉淀均呈蓝绿色,而含有突变体重组质粒pCDF-cpcB-slr2049(A24S)/slr2049(R107S)的转化菌体沉淀没有蓝绿色.镍柱纯化,SDS-PAGE检测目的蛋白与预测大小一致(图5).
将纯化的重组蛋白用紫外可见分光光度计和荧光光谱仪检测吸收光谱.从吸收光谱和荧光光谱可知,野生型slr2049 和突变体slr2049(H21S)/slr2049(L23S)/slr2049(F25S)/slr2049(W72L)/slr2049(G84S)/slr2049(Y124S)能催化PCB 正确偶联到CpcB上,产物的最大吸收峰和荧光发射峰分别在623nm 和640nm 左右,与天然产物最大吸收峰和荧光发射峰分别在625nm 和645nm 相近.而突变体slr2049(A24S)/slr2049(R107S)所催化的产物没有出现最大吸收峰和荧光发射峰.突变体slr2049(L23S)/slr2049(W72L))/slr2049(G84S)所催化的产物虽然有吸收峰,但是吸收峰处的最大值与野生型slr2049催化的产物的吸收峰值相比明显要小很多.

图5 SDS-PAGE检测纯化的pET-ho1-pcyA-cpcB-slr2049野生型和pET-ho1-pcyA-cpcB-slr2049突变体表达产物Fig.5 Examination of purified pET-ho1-pcyA-cpcB-slr2049and pET-ho1-pcyA-cpcB-slr2049mutants with SDS-PAGE
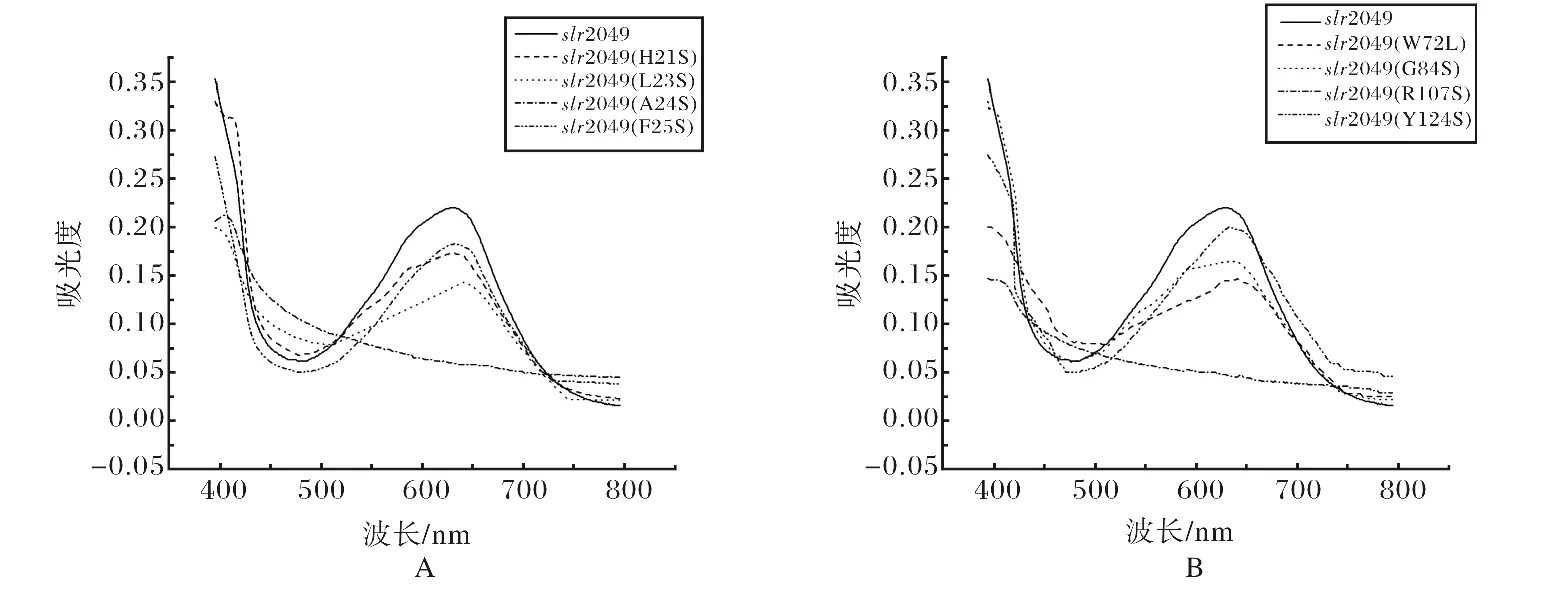
图6 体内合成的重组藻蓝蛋白β亚基紫外吸收光谱Fig.6 Absorption spectra of phycocyaninβ-subunit obtained by in vivo reconstitution
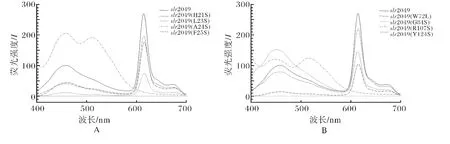
图7 体内合成的重组藻蓝蛋白β亚基荧光光谱Fig.7 Fluorescence spectra of phycocyaninβ-subunit obtained by in vivo reconstitution
3 讨论
本实验从构建突变体、表达载体、蛋白表达、SDS-PAGE凝胶电泳以及光谱扫描的结果,可以说明slr2049所编码的蛋白具有色素裂合酶的功能,同时也了解了slr2049的结构特点,以及这些结构与蛋白slr2049 在功能上的关系.光谱图上,野生型slr2049和突变体slr2049(H21S)、slr2049(L23S)、slr2049(A24S)、slr2049(F25S)、slr2049(W72L)、slr2049(G84S)、slr2049(R107S)、slr2049(Y124S)之间有光谱吸收峰值差异,说明这8个突变位点对催化生成藻蓝蛋白β亚基的能力也是有差异的.第24、107位丙氨酸和精氨酸突变所催化的表达产物在光谱图上无光谱特性,可知没有藻蓝蛋白β亚基生成;证明24、107位的丙氨酸和精氨酸是必需氨基酸,是色素裂合酶的酶活中心,这两个氨基酸的突变可以使色素裂合酶失活,丧失催化能力.第21、25、124位的组氨酸、苯丙氨酸和酪氨酸突变所催化的表达产物从光谱图可知,它们有和野生型slr2049相差不大的吸收峰,说明有藻蓝蛋白β亚基生成;证明21、25、124位的组氨酸、苯丙氨酸和酪氨酸对色素裂合酶的催化作用无明显影响,不是必需氨基酸.第23、72、84位的亮氨酸、色氨酸和甘氨酸突变所催化的表达产物从光谱图可知,它们虽然有吸收峰,但是吸收峰处的最大值与野生型slr2049最大值相比明显要小,说明有藻蓝蛋白β亚基生成,但蛋白重组效率不高;证明23、72、84位的亮氨酸、色氨酸和甘氨酸是色素裂合酶催化作用的关键位点,这3个氨基酸的突变能影响它的活性,降低它的催化能力.造成这一差异可能原因是氨基酸的定点突变能造成蛋白的结构和溶解性改变,从而影响了蛋白的催化能力.而对于其余的高保守的氨基酸位点是否是该蛋白酶的必需氨基酸和酶活性中心或是催化关键位点,待在后续的实验中进一步的研究.
藻胆蛋白的获得主要通过从藻类中提取的途径,若采用基因工程技术生产此种蛋白,必须先了解生成藻蓝蛋白的机制,才能利用基因工程技术获得大量藻胆蛋白,从而产生巨大的经济效应.唐志红等[10]、赵方庆等[11]在大肠杆菌中高效表达了蓝藻别藻蓝蛋白基因,并能大量生产高纯度重组别藻蓝蛋白——镭普克(recombinantallophycocyanin,rAPC),试验证明这一产物有良好的抗肿瘤活性和免疫能力.随着藻蓝蛋白机制的研究和基因工程技术的运用,藻胆蛋白在医药、保健、食品添加剂、试剂等方面的应用前景越来越广泛.
[1]Saune'e N A,Williams S R,Bryant D A,et al.Biogenesis of phycobiliproteins.II.CpcS-I and CpcU comprise the heterodimeric bilin lyase that attaches phycocyanobilin to Cys-82 of beta-Phycocyan-in and Cys-81of allophycocyanin subunits inSynechococcussp.PCC7002[J].J Biol Chem,2008,283:7513-7522.
[2]Scheer H,Zhao K H,Biliprotein maturation:the chromophore attachment[J].Mol Microbiol,2008,68:263-276.
[3]Shen G,Schluchter W M,Bryant D A.Biogenesis of phycobiliproteins.I.cpcS-I and cpcUmutants of the cyanobacteriumSynechococ-cussp.PCC 7002define a heterodimeric phycocaynobilin lyase specific for beta-phycocyanin and allophycocyanin subunits[J].J Biol Chem,2008,28:7503-7512.
[4]张 浩.定点突变技术的研究进展[J].免疫杂志,2000,16(4):108-110.
[5]Liu S F,Chen Y J,Lu Y D,et al.Biosynthesis of Fluorescent Cyanobacterial Allophycocyanin Trimer in Escheri-chia Coli[J].Photosynthesis Res,2010,105(2):135-142.
[6]张成武,殷志敏,欧阳平凯.藻胆蛋白的开发与利用[J].中国海洋药物,1995(3):53-54.
[7]范 晓,张士璀,秦 松,等.海洋生物技术新进展[M].北京:海洋出版社,1999.
[8]李冠武,王广策,李振刚,等.藻红蛋白介导光动力治疗的光化学机制研究[J].激光生物学报,2001,10(2):116-119.
[9]陈新美,梅兴国.螺旋藻多糖和藻胆蛋白的肿瘤防治作用及机制[J].中草药,2004,35(1):100-104.
[10]唐志红,秦 松,吴少杰,等.镭普克的制备及对小鼠H22肝癌的抑制作用[J].高技术通讯,2004,14(3):83-86.
[11]赵方庆,唐志红,林 凡,等.表达rAPC 大肠杆菌的高密度发酵及纯化产物的抑瘤活性[J].高技术通讯,2003,13(2):29-33.
