肌酸激酶测定试剂盒技术审评规范要点概述
2013-08-23郭丽丽赵阳孙嵘左霖北京市药品监督管理局医疗器械技术审评中心北京100061
郭丽丽 赵阳 孙嵘 左霖 北京市药品监督管理局医疗器械技术审评中心 (北京 100061)
2012年12 月,北京市药品监督管理局下发了关于征求肌酸激酶测定试剂(盒)产品技术审评规范意见的通知(京药监械〔2012〕93号文)。经过为期一个月的征求意见和修改补充,现已正式发布,用于指导北京市地区体外诊断试剂生产企业的注册申报工作。同时,今年该规范已立项转化为国家食品药品监督管理局2013年度产品注册审查指导原则,预计于今年年底前完成,届时将通过国家药监局向全国发布,用于指导全国范围内该产品的技术审查工作。
此外,该规范的制定作为国家863项目——心脑血管慢性损伤及急救指标等体外诊断试剂研制课题的子课题,主要目的是建立心脑血管损伤相关诊断试剂的质量评价标准,用以指导该类试剂的研制和评价工作。
心脑血管疾病是全球性的重大公共卫生问题,每年约有1700万人死于心脑血管疾病。在我国,随着人民生活水平的逐年提高,心脑血管疾病的发病率也明显升高,严重威胁人们的生命健康。作为辅助诊断心脑血管疾病指标之一的肌酸激酶,在急性心肌梗死后2~4hCK活性开始增高,可高达正常上限的10~12倍,对心肌梗死诊断较AST、LDH特异性高。目前国内外众多临床化学体外诊断试剂生产厂家均开发了此项目,截止2013年3月31日,全国共有有效注册证约125个(含国产、进口)。根据前期调研发现,该产品名称多样、性
能及技术指标不统一,缺乏统一的评价标准,为了规范该产品的注册申报及技术审评工作,特制定本规范。下面就规范中的重点问题进行简要概述。
1.适用范围
该规范适用于采用分光光度法原理,利用全自动、半自动仪器或分光光度计,在医学实验室进行肌酸激酶定量检验所使用的临床化学体外诊断试剂(盒)。干式肌酸激酶测定试剂(盒)如胶体金免疫层析法的产品不在本文讨论范围之内。
2.试剂盒命名、组成及用途
2.1 试剂盒命名
试剂(盒)名称由三部分组成:第一部分 被测物名称:肌酸激酶;第二部分 用途;测定试剂(盒);第三部分 方法或原理:磷酸肌酸底物法。
规范制定之前产品申报名称方法学不统一,如有肌酸激酶试剂盒(酶偶联法)、肌酸激酶试剂盒(IFCC推荐法)、肌酸激酶试剂盒(速率法),现根据临床化学体外诊断试剂(盒)命名行业标准报批稿对产品名称进行了统一规范。
2.2 反应原理
CK催化CrP转变成Cr,同时ADP磷酸化成ATP,然后进行酶偶联反应。反应式如下:
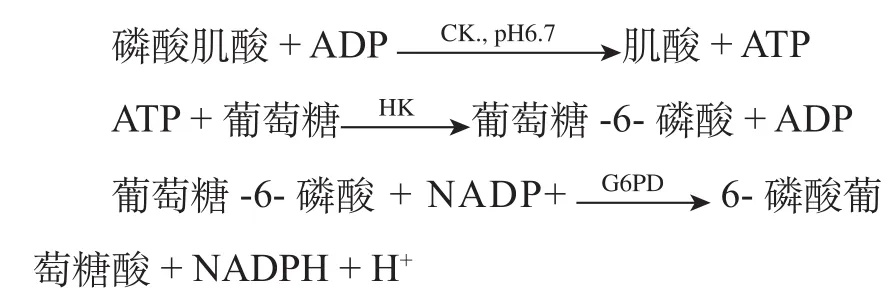
上述反应式中,第一步CK反应产生ATP,用HK/G6PD(己糖激酶/葡萄糖-6-磷酸脱氢酶)偶联反应,最终使NADP转变成NADPH,可在340nm波长进行监测。
2.3 产品适用的相关标准
(1)GB/T 191-2008 包装储运图示标志;
(2)GB/T 21415—2008 体外诊断医疗器械 生物样品中量的测量 校准品和控制物质赋值的计量学溯源性
(3)GB/T 26124-2011 临床化学体外诊断试剂(盒)
(4)YY/T 0316-2008 医疗器械 风险管理对医疗器械的应用;
(5)YY 0466-2003 医疗器械 用于医疗器械标签、标记和提供信息的符号;
(6)YY/T 0638—2008 体外诊断医疗器械 生物样品中量的测量 校准品和控制物质物中酶催化浓度赋值的计量学溯源性。
2.4 产品的预期用途
本试剂(盒)用于体外定量测定人血清或血浆中的肌酸激酶的活性。
注:需要注意的是,在产品注册过程中,如果企业宣称的预期用途中可适用不同的样本类型,均应经过临床验证。
3.产品的主要技术指标
本产品的性能指标主要参照《肌酸激酶测定试剂(盒)》行业标准报批稿、GB/T26124-2011《临床化学体外诊断试剂(盒)》制定及已发布的北京药监局技术审评规范《临床化学体外诊断试剂(盒)产品技术审评规范》。
3.1 外观
目测检查,符合生产企业规定的正常外观要求。
一般要求试剂无杂质、无絮状物,外包装完整无破损。如有校准品或干粉试剂,应明确每个组份的外观要求。
3.2 净含量
用通用量具测量,液体试剂的净含量应不少于标示值。
3.3 试剂空白
3.3.1 试剂空白吸光度
用指定空白样品测试试剂(盒),在测试主波长下,37℃、340nm波长、1cm光径条件下,记录测试启动时的吸光度(A1)和约5分钟(T)后的吸光度(A2),A2测试结果即为试剂空白吸光度测定值,应不大于0.50。
3.3.2 试剂空白吸光度变化率

3.4 分析灵敏度
用已知浓度或活性的样品进行测试,记录在试剂(盒)规定参数下产生的吸光度改变。换算为n单位吸光度差值(∆A)或吸光度变化(∆A/min)。应符合生产企业给定范围。
注:分析灵敏度是指单位浓度的样本引起吸光度变化的程度,用以反应试剂对样本的响应程度,建议厂家给出样本浓度,并给出该浓度下吸光度变化的范围。
3.5 线性范围
用超出线性范围上限活性的样品和超出或等于线性范围下限活性的样品,混合成至少5个稀释浓度(xi)。分别测试试剂盒,每个稀释浓度测试3次,分别求出测定结果的均值(yi)。以稀释浓度(xi)为自变量,以测定结果均值(yi)为因变量求出线性回归方程。按公式(1)计算线性回归的相关系数(r)。
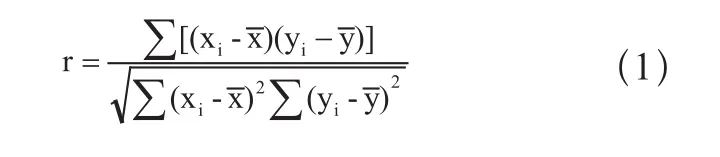
稀释浓度(xi)代入求出线性回归方程,计算yi的估计值及yi与估计值的相对偏差或绝对偏差。
试剂盒至少在(25~1000)U/L(37℃)的线性范围内分析性能应符合如下要求:
3.5.1 线性相关系数r应≥0.990;
3.5.2 (25~100]U/L 范围内,线性绝对偏差应不大于±10 U/L;(100~1000)U/L 范围内,线性相对偏差应不大于±10%。
3.6 重复性
3.6.1 批内重复性
在重复性条件下,分别用(35±10)U/L、(200±20) U/L和 (900±100)U/L的血清样品或质控样品测试试剂(盒),重复测试至少10次(n≥10),分别计算测量值的平均值(x)和标准差(s)。计算变异系数(CV),应不大于5%。
注:重复性所使用样本浓度的设定考虑了该检验项目的参考值区间,选择在参考范围内以及参考范围外不同浓度进行验证。
出厂检验时生产企业可以根据产品特性对两个浓度样品进行检验。
3.6.2 批内瓶间差(干粉或冻干试剂适用)
分 别 用(35±10)U/L 、(200±20) U/L和(900±100)U/L的血清样品或质控样品测试同一批号的10个待检试剂(盒),并计算每个浓度10个测量值的平均值(x1)和标准差(s1)。
分 别 用(35±10)U/L 、(200±20) U/L和(900±100)U/L的血清样品或质控样品对该批号的1个待检试剂(盒)重复测试10次,计算每个浓度结果的均值( 2)和标准差(s2)按公式(2)、(3)计算瓶间差的变异系数(CV)。
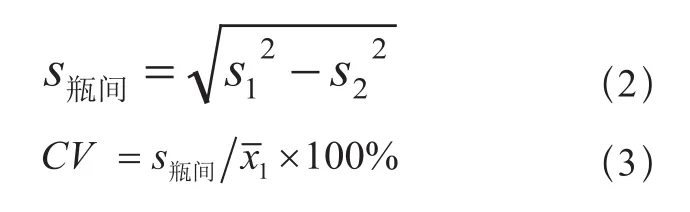
当s1<s2时,令CV=0
每个浓度下试剂(盒)批内瓶间差均应不大于5%。
3.6.3 批间差
用(200±20) U/L的血清样品或质控样品分别测试3个不同批号的试剂(盒),每个批号测试3次,分别计算每批3次测定的均值(i=1,2,3),按公式(4)、(5)计算相对极差(R)。

式中:
–中的最大值 ;
中的最小值。
试剂(盒)批间差相对极差应不大于10%。
3.7 准确度
3.7.1 相对偏差
3.7.1.1 用可用评价常规方法的有证参考物质(CRM)对试剂(盒)进行测试,重复检测3次,取测试结果均值(M),按公式(6)计算相对偏差(B%)。相对偏差应不大于10%。
3.7.1.2 用由IFCC肌酸激酶参考测量程序定值的参考范围上限和参考范围上限2至5倍浓度水平各一个人源样品(可适当添加被测物,以获得高浓度的样品)对试剂(盒)进行测试,每个浓度样品重复检测3次,分别取测试结果均值,按公式(6)计算相对偏差。相对偏差应不大于10%。

式中:M为测试结果均值;
T为标准物质标示值,或各浓度人血清定值。
3.7.2 比对试验
参照CLSI EP9-A2的方法,用不少于40个在检测浓度范围内不同浓度的人源样品,以生产企业指定的分析系统作为比对方法,每份样品按待测试剂(盒)操作方法及比对方法分别检测。用线性回归方法计算两组结果的相关系数(r)及每个浓度点的相对偏差。相关系数 r≥0.975,相对偏差应不大于±10%
注:1.型式试验时,可采用上述方法之一测试试剂(盒)的准确度。
2.出厂检验时,生产企业可以选择以上检验方式,或用与选定校准品配套的质控品进行准确性实验,试验方法自行确定。
根据企业实际质量控制的情况,出厂检验可采用校准品或选定的配套质控品进行检验,但企业应每年应以型式检验的方式对产品的准确度进行验证。
3.8 稳定性
3.8.1 效期稳定性:试剂(盒)在规定的贮存条件下保存至有效期末,产品的性能应至少符合线性、准确度和重复性的要求。
3.8.2 热稳定性试验:取有效期内样品根据生产企业声称的热稳定性条件,产品的性能应至少符合线性、准确度和重复性的要求。
3.8.3 复溶稳定性(干粉或冻干试剂适用):干粉试剂开瓶后(复溶后)在规定的贮存条件下保存至预期时间内,产品的性能应至少符合线性、准确度和重复性的要求。
3.9 校准品和质控品(如适用)
若试剂盒配套校准品和质控品,应提供校准品溯源性说明及质控品定值说明。
注:校准品溯源应参照GB/T 21415—2008 《体外诊断医疗器械 生物样品中量的测量 校准品和控制物质赋值的计量学溯源性的要求》给出列出溯源图,并描述不确定度的计算方法和限制范围。质控品定值应采用不同实验室赋值的方式。
4.产品的临床要求
试剂(盒)按照《体外诊断试剂注册管理办法》(试行)、《体外诊断试剂临床研究技术指导原则》及《北京市第二类体外诊断试剂临床试验指导原则》进行临床试验。执行的基本原则至少包括以下几个方面:
4.1 在两家省级医疗卫生单位完成临床研究;
4.2 “已有同种批准上市”的试剂(盒)应与已批准上市产品针对临床样本进行比对试验。
4.3 临床样本的要求:综合不同地区人种、流行病学背景、病原微生物的特性等因素选择研究单位,症状典型和非典型的,一般临床研究的总样本数至少为200例。
4.4 如果申报的试剂产品同时适用于血清、血浆样本。血清(或血浆)的试验例数按照《体外诊断试剂临床研究技术指导原则》要求的例数进行;同时,还要进行血清、血浆检验结果一致性的比对。
4.5 异常样本、正常样本应均匀分布,异常样本量应不少于30%。
5.试剂(盒)说明书、标签、包装标识
5.1 试剂(盒)说明书
试剂(盒)说明书应当符合《体外诊断试剂说明书编写指导原则》的要求,需要注意以下内容:
(1)产品名称
通用名称:肌酸激酶测定试剂盒(磷酸肌酸底物法)
商品名称:与企业声称一致。
英文名称:Creatine kinase Assay Kit
(2)应说明试剂(盒)的包装规格;
(3)预期用途:本试剂(盒)用于体外定量测定人血清或血浆中的肌酸激酶的活性。
(4)临床意义:肌酸激酶主要存在于骨骼肌和心肌,在脑组织中也存在。各种类型的进行性肌萎缩时,血清CK活性增高。神经因素引起的肌萎缩如脊髓灰白质炎时,CK活性正常。皮肌炎时CK活性可轻度或中度增高。急性心肌梗死后的2~4hCK活性开始增高,可高达正常上限的10~20倍。CK对心肌梗死诊断较AST、LDH的特异性高。但此酶增高的持续时间较短,24~天就恢复正常。病毒性心肌炎时。CK活性也明显升高,对诊断及判断预后有参考价值。
CK增高还见于脑血管意外、脑膜炎、甲状腺功能低下患者。还应注意到,一些非疾病因素如剧烈运动、各种插管及手术、肌内注射冬眠灵和抗生素也可能引起CK活性增高。
注:生产企业可根据自身产品情况编写,如有新的临床意义,应提供相应依据。
(5)检验原理:
CK催化CrP转变成Cr,同时ADP磷酸化成ATP,然后进行酶偶联反应。反应式如下:
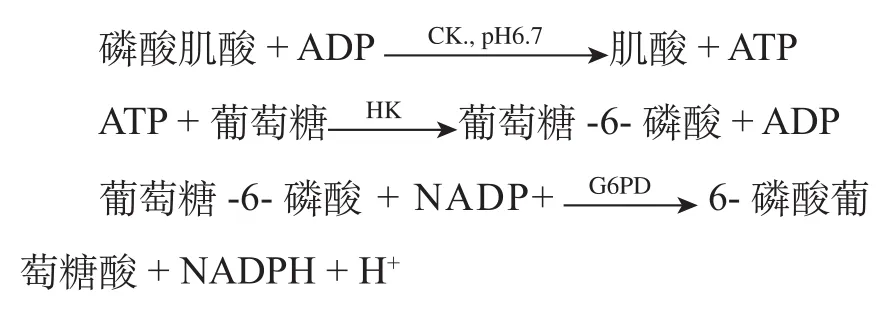
上述反应式中,第一步CK反应产生ATP,用HK/G6PD(己糖激酶/葡萄糖-6-磷酸脱氢酶)偶联反应,最终使NADP转变成NADPH,可在340nm波长进行监测。
(6)主要组成成分:对于产品中包含的试剂组份应有名称、数量、每一组份中的主要成分在反应体系中的比例或浓度,如果对于正确的操作很重要、应提供其生物学来源、活性及其他特性;对于试剂盒中含有校准品、质控品,说明主要组成成份及其生物学来源,注明校准品的定值及其溯源性,注明质控品的允许范围。
(7)应说明贮存条件及有效期;
(8)应说明可适用仪器的名称及型号;
(9)应说明对样本的要求,包括样本采集过程中的注意事项,为保证样本各组分稳定所必需的抗凝剂或保护剂,已知的干扰物、能够保证样本稳定的贮存、处理和运输的方法;
如:血清样本室温保存4h、4℃保存 8~12h。标本采集后应尽快分离出血清,避光至冰箱保存。样本保存期限应提供依据。
(10)应说明检验方法,至少包括试剂配制、试验条件、校准程序、校准间隔、质量控制程序、试验结果的计算;
(11)应有参考值范围,并简要说明确定的依据;
如: 健康成年男性:38~174U/L;
健康成年女性:26~140U/L。
参考值范围参考《全国临床检验操作规程》(第3版),并经过本公司对120临床样本验证获得,各临床实验室应建立自己的参考范围区间。
(12)应有检验结果的解释,说明可能对试验结果产生影响的因素;
(13)应说明检验方法的局限性;
说明该检验方法由于哪些原因会使测量结果产生偏离。
如:红细胞不含CK,轻度溶血无影响。中度及重度溶血时,因红细胞释放出AK、ATP、及G6PD会干扰检测结果。
若超出线性范围可稀释后测定,应明确最大稀释比例。
(14)应说明产品性能指标,并符合注册产品标准的要求;
(15)应说明必要的注意事项,如本品仅用于体外诊断;CK活性不稳定,血清保存期间易丧失活性,应严格按照样本要求操作等,若该产品含有人源或动物源物质,应给出具有潜在感染性的警告;
(16)注明引用的参考文献;
(17)注明生产企业,包括企业名称、生产/注册地址、联系方式;
(18)注明医疗器械生产企业许可证编号;
(19)注明医疗器械注册证书编号;
(20)注明产品标准编号;
5.2 外包装标签
(1)生产企业名称和地址。
(2)试剂(盒)名称。
(3)批号。如试剂(盒)包含不同批号的组件,外包装的批号应能保证每个组件的批号可从生产企业的生产记录中溯源。
(4)规格。应包含体积或复溶后的体积。
(5)预期用途。如试剂(盒)名称不能反映试剂(盒)的预期用途,应提供简要的预期用途说明。
(6)体外诊断用途。应说明试剂的体外诊断用途。
(7)储存和处置条件。应提供在未开封状态下可保证试剂、校准品和质控物质的稳定状态的必要储存条件;应规定影响稳定性的其他条件(如适用);应规定产品处置时所采取的所有其他特殊措施(如适用)。
(8)失效期。应明示在规定储存条件下的失效期;失效期应以年、月,适当时以日表示;如仅给出年月,失效期应为指定月的最后一天;外包装标签上明示的失效期应为最早到期组件的失效期。
(9)警告和预防措施。如体外诊断试剂(盒)被认为有危险性(例如:化学,放射性或生物危害性),外包装应标有适当的警示危险的文字或符号,YY/T 0316的要求适用;对于化学危害,如试剂(盒)没有随带含有适当的危险和安全性说明的使用说明,则应在外包装的标签上进行说明。
5.3 初始包装标签
如初始包装同时也是外包装,则外包装标签的要求也适用。
初始包装标签上应提供如下信息:
小标签规定。如瓶标签上可利用的空间太小以致于不能包括下述所需全部信息,则5)、6)和7)项的信息可省略或删除。
(1)生产企业名称或等同的商标或标志。
(2)产品名称。产品名称应确保使用者能正确识别产品。
(3)批号。
(4)规格。例如:质量,体积,复溶后的体积。
(5)体外诊断用途。
(6)储存和处置条件。应提供未开封状态下保证产品的稳定状态必需的储存条件;如与外包装提供的条件不同,还应提供产品处置所采取的所有其他特殊措施;
(7)失效期。应明示规定储存条件下的失效期,表示方式见外包装标签。
(8)警告和预防措施。如体外诊断试剂(盒)被认为有危险性(例如:化学,放射性或生物危害性),外包装应标有适当的警示危险的文字或符号,YY/T 0316的要求适用;对于化学危害,如试剂(盒)没有随带含有适当的危险和安全性的说明的使用说明,则应在外包装的标签上进行说明;适用时,应明示试剂预期为一次性使用。
6 .其他相关要求
6.1 注册单元划分的原则
试剂(盒)的注册单元应为单一试剂(盒),一个注册单元可以包括不同的包装规格。
6.2 出厂检验原则
每批试剂(盒)出厂检验至少进行以下几项内容:外观、装量、试剂空白吸光度或吸光度变化率、准确度、批内重复性。
7.结语
本规范主要依据《医疗器械监督管理条例》、《体外诊断试剂注册管理办法》(试行)、《体外诊断试剂临床研究技术指导原则》、《体外诊断试剂说明书编写指导原则》进行编写。产品的主要技术指标参考了GB/T 26124-2011 临床化学体外诊断试剂(盒)及肌酸激酶国家标准的报批稿,在规范的制订过程中我们征求了如北京医院、安贞医院、煤炭总医院等众多临床检验领域的专家意见,还征求了包括北京九强、利德曼、中生北控等生产企业的意见。
本文所介绍的规范已经用于指导和规范北京市肌酸激酶产品的技术审评工作,帮助审评人员更深入的理解和掌握该类产品的反应原理、性能指标、包装标识等内容,把握技术审评工作的基本要求和尺度,对产品安全性、有效性作出更全面的系统评价。同时,规范的实施也更有利于指导生产企业的注册申报工作,促进产品安全有效上市。
[1] 《体外诊断试剂分析性能评估(准确度-方法学比对)技术审查指导原则》
[2] GB/T 26124-2011《临床化学体外诊断试剂(盒)》
[3] 《体外诊断试剂说明书指导原则》
[4] 《北京市第二类体外诊断试剂临床试验指导原则》
[5] 《全国临床检验操作规程》(第三版)
