相转移催化氧化燃料油脱硫技术研究进展
2013-08-20赵田红
赵田红,张 燕
(西南石油大学化学化工学院,成都 610500)
燃料油的传统脱硫技术主要分为两大类:加氢脱硫和非加氢脱硫。加氢脱硫是工业燃料油脱硫的主要手段,可脱除燃料油中硫醇、硫醚等简单小分子有机硫,具有脱硫率高、燃料油收率高等优点,但在脱除燃料油中含量很高的苯并噻吩(BT)、二苯并噻吩(DBT)及其衍生物时,不仅需高温高压等较苛刻条件[1],还需专门的高效催化剂和大量高纯度的氢气,加大了工艺设备投资和操作费用;另外,深度加氢脱硫过程中会降低烯烃和芳香烃的含量,从而大大降低燃料油的辛烷值[2]。而非加氢脱硫在常温常压下即可进行,操作条件温和,不需氢气,操作和投资费用较低,环境污染少,自世界各国制定新标准降低燃料油中硫含量后,燃料油的深度脱硫技术越来越受到人们的关注,尤其是氧化脱硫技术[3]。
氧化脱硫作为一种新的燃料油深度脱硫技术,因其投资少、操作费用低受到国内外学者的重视。氧化脱硫采用的氧化剂主要是H2O2,对加氢脱硫技术难以脱除的噻吩类及其衍生物有很好的脱除效果,使燃料油中硫含量进一步降低;另一方面其氧化产物是水,不会对环境产生任何污染。然而,氧化脱硫是在两相系统中进行的,水相的氧化剂和油相的含硫化合物接触面积小、相间阻力大,导致传质速率低,造成氧化剂与含硫化合物反应较困难,从而影响氧化反应速率和脱硫效果。相转移催化剂的应用不仅能使反应在相对温和的条件下进行[4],还能有效地增加氧化剂在油相的分布,增大两相接触面积,减小反应阻力,加快反应速率,提高脱硫效果。
1 有机酸-相转移催化氧化燃料油脱硫技术
在有机酸体系中,相转移催化氧化燃料油脱硫的反应过程一般包括2个阶段:一是有机酸和氧化剂结合形成氧化活性组分RCOO-,相转移催化剂阳离子Q+和氧化活性组分RCOO-在水相或相界面处结合形成离子对 Q+RCOO-,Q+就将RCOO-从水相“萃取”至有机相中;二是在有机相中,RCOO-和含硫化合物进行有效接触并发生氧化反应,将含硫化合物氧化成砜类或亚砜类。RCOO-和Q+又重新组成离子对Q+RCOO-返回水相,在水相中进行下一个循环的反应,其催化机理见图 1[5]。
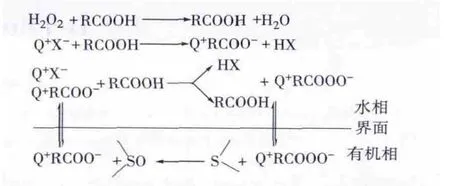
图1 有机酸-相转移催化氧化脱硫机理
赵地顺等[6-8]在H2O2/有机酸体系中进行了一系列相转移催化油品脱硫的研究,并取得了一些有意义的成果。当汽油与氧化体系体积比为0.2∶1,双氧水与乙酸酐体积比为 5∶1,十六烷基三甲基溴化铵(CTAB)用量为0.06 g,FCC汽油反应 0.5 h,脱硫率可达 80.13%;在 H2O2/甲酸(物质的量比1∶1)体系中,加入四丁基溴化铵(TBAB)作为相转移催化剂对噻吩溶于正庚烷的轻质油品进行脱硫实验,40℃时反应1.5 h,则脱硫率达86.36%;若TBAB为相转移催化剂,H2O2为氧化剂,乙酸酐为助氧化剂,采用选择性氧化法对FCC汽油进行脱硫研究,当汽油与氧化体系体积比为1∶5,双氧水与乙酸酐体积比为1∶1,TBAB为0.02 g,室温下反应0.5 h时,FCC 汽油的脱硫率达 92.10%,收率为 90.3%。
Komintarachat 等[9]采用[(C4H9)4N]2W6O19,[(C4H9)4N]4V(VW11)O40和[(C4H9)4N]4PVW11O40为催化剂,在H2O2/CH3COOH体系中,进行了一系列催化氧化轻质油脱硫的实验。结果表明,当n(氧化剂)∶n(模型油)=100∶1,n(模型油)∶n(催化剂)=50∶1,n(H2O2)∶n(CH3HCOOH)=1∶1,反应温度60 ℃,搅拌反应5h,以[(C4H9)4N]4V(VW11)O40为相转移催化剂的脱硫效果最好,可使柴油的硫含量(质量分数,下同)从0.575%降至0.055%,脱硫率高达90.4%。
赵立明等[10]以 H2O2/HCOOH 为氧化剂、TBAB为相转移催化剂,当水相中TBAB含量为0.2%,H2O2含量为16%,HCOOH含量为8%、剂油比为3.10时,70℃条件下高速搅拌反应45 min,噻吩脱除率达89%以上。佘林源等[11]发现在以上催化氧化体系中,反应物比例不同,也会大大影响脱硫效果。在反应条件:w(TBAB)=0.10%,V(H2O2)∶V(柴油)=1∶10,V(H2O2)∶(HCOOH)=1∶2,剂油体积比 2∶5,搅拌速度为150 r/min,反应温度60℃,反应时间2 h,直馏柴油的脱硫率达82.03%,收率为92.02%。
Zhao等[12]在 H2O2/HCOOH 氧化体系中,以四丁基硫酸氢铵(TBSB)为相转移催化剂,在最佳反应条件下,模型油中 DBT的脱硫率高达97.45%;将此催化氧化体系应用于实际FCC汽油脱硫时,脱硫率为88.34%,也显示了良好的脱硫效果。
张海燕等[13]以十八烷基甜菜碱[C18H37N+(CH3)2CH2COO-]为相转移催化剂,在乳液体系中对成品汽油和催化裂化汽油中硫化物分别进行催化氧化实验,在最佳工艺条件下:m(剂)∶m(油)=0.01∶1,n(氧化剂)∶n(催化剂)=3∶4,常温下反应30 min,成品汽油的硫含量最低可降至39 μg/g,脱硫率为84%;FCC汽油的硫含量最低降至 36 μg/g,脱硫率为 88.4%。
综上所述,在H2O2/有机酸氧化体系中加入相转移催化剂,脱硫效果明显提高。过氧化氢与有机酸反应,可生成氧化性更强的过氧酸,过氧酸可使燃料油中非极性有机硫化物转化成极性硫化物——砜类,相转移催化剂可使界面张力达最佳值,增加相界面积,提高反应速率。相转移催化剂的加入缩短了反应时间、增加了脱硫效果等,但还存在氧化剂选择性差、油品收率偏低等问题。因此,寻求催化活性更高的相转移催化剂是H2O2/有机酸氧化体系发展的方向。
2 杂多酸-相转移催化氧化燃料油脱硫技术
杂多酸与季铵盐离子对结合成相转移催化剂一般有2种形式:1)杂多酸(H+Y-)与季铵盐(Q+X-)以混合物形式作为催化剂,进入体系后结合成离子对,与氧化剂作用生成Q+Y-[O],Q+Y-[O]与含硫化合物发生氧化反应,脱除硫。2)杂多酸阴离子Y-和季铵盐阳离子Q+以离子对的形式存在于反应中,季铵盐阳离子是亲油基团,可将不溶于油相的杂多酸阴离子带入油相中,发挥催化作用。1)和2)中的催化作用机理如图2 所示[14]。
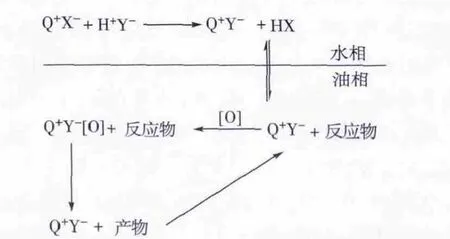
图2 杂多酸(H+Y-)/季铵盐(Q+X-)以混合物的形式为催化剂-相转移催化氧化脱硫机理
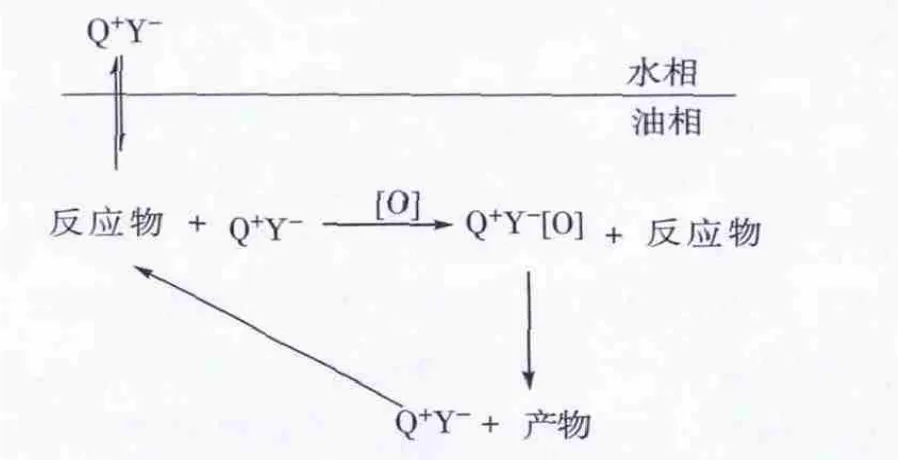
图3 杂多酸Y-和季铵盐Q+以离子对形式为催化剂-相转移催化氧化脱硫机理
Huang 等[15]在 H2O2/磷钨酸氧化体系中加入一系列不同碳链长度的季铵盐(C12,C14,C16,C18)作为相转移催化剂进行DBT脱硫实验的研究。结果表明,DBT的氧化脱硫效果随着季铵盐碳链增长而明显增强;当反应温度为50℃,反应时间20 min,季铵盐和磷钨酸物质的量比为1∶1时,能完全氧化模拟油中硫含量为3 000 μg/g的二苯并噻吩中的硫化物。
张予辉等[16]将磷钨杂多酸季铵盐相转移催化剂/H2O2(Q3[PO4(WO3)4]/H2O2)体系应用于FCC汽油的液-液高效催化氧化降低烯烃含量的实验中,在V(H2O2)=2.5 mL,剂油质量比为1∶40,pH 为3.3,反应温度60 ℃,反应时间1 h的条件下,FCC汽油烯烃体积分数下降了23.56%,而汽油辛烷值几乎没变,处理后的FCC汽油完全符合我国清洁汽油规定的烯烃体积分数低于35%的新标准。
周二鹏等[17]用H2O2/磷钨酸氧化模型含硫化合物,加入TBAB相转移催化剂,氧化剂用量为n(H2O2)∶n(S)=4∶1,反应温度40 ℃,反应时间150 min,硫含量从 500 μg/g 降至 34.5 μg/g,脱硫率为93.1%。
He等[18]系统研究了短烷基链的过氧磷钼酸和离子液体对H2O2氧化脱硫效果的影响。结果表明,以[(C4H9)4N]3{PO4[MOO(O2)2]4}为催化剂,使用1-丁基-3-甲基咪唑四氟硼酸盐([BMIM]BF4)离子液体,脱硫效果最好,DBT脱硫率最高达97.3%。另外,不使用离子液体时,H2O2/[(C4H9)4N]3{PO4[MOO(O2)2]4}、H2O2/[C14H29N(CH3)3]3{PO4[MOO(O2)2]4}和H2O2/[C16H33NC5H5]3{PO4[MOO(O2)2]4}的脱硫率均低于20%;加入1-丁基-3-甲基咪唑四氟硼酸([BMIM]BF4)离子液体后,脱硫率均大幅提高,且碳链短的季铵盐相转移催化剂比长碳链的催化活性好。
介质消耗中大部分是产品和磁选机尾矿带走的介质损失。选煤厂设计规范中规定重介质选煤吨原煤的磁铁矿粉损耗量,块煤系统小于0.8 kg/t原煤;末煤系统小于2.0 kg/t原煤。
邱江华等[19]以十八烷基三甲基磷钼酸铵为相转移催化剂,H2O2为氧化剂,对模型油进行氧化脱硫研究,70℃时反应150 min,结果表明,DBT和BT的脱除率分别高达100%和40.5%;在相同条件下,磷钼酸季铵盐作相转移催化剂时,DBT和BT的脱除率分别是磷钼酸作相转移催化剂时的6.3倍和3.4倍;同样条件下,当反应时间延长至3 h,磷钼酸季铵盐用于直馏柴油的脱硫时,柴油的脱硫率达88.7%,收率不低于99%。
刘帅等[20]以[(C16H33)N+(CH3)3]3[PMO12O40]-/H2O2双亲催化氧化体系结合萃取对柴油中含硫化合物进行脱硫研究,最佳反应条件:H2O2与S物质的量比为5∶1,反应温度80℃,反应时间90 min,以γ-丁内酯萃取时,柴油的脱硫率和收率分别为87.8%和86.0%。
徐亚荣等[21]以 CTAB为相转移剂,采用H2O2/磷钨酸催化氧化体系对乌鲁木齐石化公司烃重组富芳汽油进行氧化脱硫研究,在相转移催化剂加量(以H2O2质量计,下同)0.5%,杂多酸加量1%,反应温度50℃,反应时间3 h,汽油脱硫率高达81.95%,收率不低于97%。
蔡永宏等[22]制备了催化剂(BMIM)2(CTMA)PMO12O40(二-(1-丁基-3-甲基咪唑)-十六烷基三甲基磷钼酸铵盐),在n(C)∶n(S)=0.06∶1,n(H2O2)∶n(S)=6∶1,V(乙腈)=5 mL,反应温度60℃,反应时间60 min的条件下,使模拟汽油的脱硫率达69.4%,同样条件下用于真实汽油的脱硫,则将真实汽油硫含量从原来的196.8 μg/g降至 61.4 μg/g,脱硫率为68.8%。
Zhang 等[23]系统研究了[π -C5H5NC16H33]3[PW4O16],H3PW12O40和[(C4H9)4N]3[PW12O40]与氧化剂H2O2一起氧化3-甲基噻吩(3-MT)、BT和 DBT的脱硫实验,结果表明,[π-C5H5NC16H33]3[PW4O16]因具有在油品中较好的分散性和硫化物吸附性,其脱除BT和DBT的效果优于其他两种相转移催化剂,在1.4 MPa O2、90℃的温和条件下,反应时间6 h,可将96%的硫化物转化成砜,再通过沉淀或过滤等方法去除反应产物中的砜。
杂多酸是一种固体酸催化剂,可溶解在极性较强的小分子溶剂中,用于均相或多相催化反应;在燃料油氧化脱硫过程中具有更高的催化活性和选择性,增加了氧化剂的利用率;通过将杂多酸催化剂负载在载体和金属上改性,可进一步提高其催化活性和选择性。但季铵盐等相转移催化剂不能直接从燃料油中分离,无法循环利用,对油品性能和生产成本均有一定的影响。
3 离子液体-相转移催化氧化燃料油脱硫技术
离子液体相转移催化剂和传统相转移催化剂的催化机理有所不同,传统相转移催化剂在反应过程中需加入除反应物之外的第三种物质,而离子液体则不需再加入第三种物质,其既可作为反应物或溶剂,也可作为相转移催化剂。在反应过程中,离子液体以一种反应物形式参与反应,当反应结束后则以相转移催化剂的形式返回非反应相,实现反应的循环进行。
Zhang等[25]研究了酸性离子液体N-丁基-N-甲基咪唑硫酸氢盐([BMIM]HSO4)的用量和循环使用次数对油品脱硫效果的影响。以H2O2为氧化剂,DBT溶于正辛烷为模型油,V(oil)=5 mL,m(IL)=3 g,n(O)∶n(S)=4∶1,反应温度60 ℃,氧化反应时间60 min,模型油的脱硫率高达100%,离子液体循环利用5次,脱硫率未见明显下降。
曹群等[26]利用离子液的合成原理将乙酸嫁接在甲基咪唑上合成乙酸型离子液,使离子液体同时具有相转移催化和萃取的功能,在反应温度80℃,反应时间30 min,当模拟油、离子液体和H2O2等体积时,表现出最佳的脱硫效果,模拟油中的噻吩脱除率达到73%。在这个实验中,催化剂乙酸被固定在甲基咪唑上,避免了乙酸在挥发性实验中极易损失并易进入到油相中的问题,减少了乙酸的挥发和毒性,有利于其发挥催化功能。
Karolina等[27]把将1-丁基 -3-甲基咪唑三氟甲磺酸盐([BMIM][OTf])离子液体应用在模型汽油和模型柴油的脱硫实验中,在萃取温度298.15 K、大气压力下,经过1 h的萃取,[BMIM][OTf]离子液体能萃取出模型汽油中96%的噻吩;且[BMIM][OTf]离子液体可脱除模型柴油中97.5%的二苯并噻吩和93%以上的噻吩。
近年来,离子液体越来越广泛地应用于燃料油氧化脱硫技术当中,利用噻吩衍生物与离子液体在室温下形成配合物的性质可以实现油品的深度脱硫。离子液体可以循环利用多次,具有较好的应用前景,但是离子液体成本较高,目前仍处于实验室研究阶段,未能实现工业化。
4 其他
Cheng等[28]以咪唑型离子液体为相转移催化剂进行海军柴油(F-76)的深度脱硫实验,30%H2O2和20%三氟乙酸为氧化剂,超声波辅助氧化10 min,在50℃时搅拌反应170 min,可完全氧化海军柴油中的含硫化合物,实现了100%脱硫。
离子液体和超声波结合进行油品的催化氧化脱硫,综合了超声波和离子液体的优点,不仅具有超声波缩短反应时间的特点,还具有离子液体与含硫化合物形成配合物进行深度脱硫的特点,大大提高了脱硫效率,甚至可实现完全脱硫。但由于生产大功率密度超声设备存在一定困难,因此该技术不仅设备投资费用高,且难于实现工业化。
赵地顺等[29]采用光催化氧化与液液萃取同时进行的方法,以CTAB为光敏剂,H2O2为氧化剂,加入30 mL FCC汽油、90 mL双氧水和0.20 g光敏剂反应,在pH为5、365 nm的300 W中压汞灯照射10 h下高速搅拌5 min,FCC汽油脱硫率达91.20%,且脱硫后双氧水和光敏剂可重复使用,不会造成二次污染。
Yasuhiro Shiraishi等[30]使用 H2O2和 3 倍的光敏剂二苯甲酮(BZP),氧化脱除油品中DBT,使因芳烃存在而被抑制的脱硫效果大大提高。反应体系中同时加入BZP和H2O2,在大于280 nm的光照射48 h下,最后利用固体吸附剂从过氧化氢水溶液中分离出光氧化后的含硫化合物,经分析可将商业轻质油中的二苯并噻吩含量从0.20%降至0.05%,符合日本对燃料油硫含量新法规的要求;另外,在波长大于280 nm的光辐射下,氧化系统中同时使用H2O2和BZP脱除C5和C6烷基取代DBT的效果远高于单独使用H2O2或BZP的脱硫效果,但此方法不能有效地脱除直馏轻柴油中的DBT,只适用商业轻质油品的脱硫。
在光催化氧化与液液萃取结合催化氧化燃料油脱硫中,光敏剂不仅是光引发剂,还可作为相转移剂,在光催化氧化反应时具有双重作用,因而脱硫效果好,且具有经济、环保的优点,此方法将在燃料油脱硫技术具有比较广阔的发展前景。
5 结语
1)H2O2氧化脱硫具有反应条件温和、选择性好、对原料适应能力强、脱硫率高等特点,是目前氧化脱硫方法中研究最多的一种方法。但H2O2氧化脱硫体系普遍存在油水两相接触不充分、氧化剂浪费等缺点,将相转移催化与氧化脱硫结合,能克服非均相传质困难、氧化剂未充分发挥氧化作用等问题,不仅缩短了反应时间,还降低了能耗和操作成本。
2)相转移催化与超声波、光辐射、离子液体等相结合方法表现出了更好的脱硫效果,因此,研究范围应扩展到多种脱硫方法相结合的领域,使相转移催化氧化脱硫技术尽快适应大规模工业化生产。
[1]Song C S.An overview of new approaches to deep desulfuriza-tion for ultra-clean gasoline,diesel fuel and jet fuel[J].Catalysis Today,2003,86(1/2/3/4):211-263.
[2]曾丹林,胡义,王可苗,等.燃料油萃取脱硫技术研究进展[J].石油炼制与化工,2012,43(5):98-102.
[3]路文娟,杨延钊.过氧化氢用于油品氧化脱硫的研究进展[J].化工进展,2009,28(4):605-609.
[4]李倩.杂多酸/季铵盐体系在燃料油催化氧化脱硫中的应用[J].技术纵横,2012,31(1):70-71.
[5] 赵地顺.相转移催化原理及应用[M].北京:化学工业出版社,2007:208-209.
[6]赵地顺,马四国,刘翠微,等.相转移催化应用于催化裂化汽油氧化脱硫的研究[J].高等学校化学学报,2006,27(1):144-146.
[7]赵地顺,任红威,李乐.季铵盐相转移催化氧化噻吩脱硫的研究[J].高等学校化学学报,2007,28(4):739-742.
[8]赵地顺,马四国,刘翠微.FCC汽油选择性氧化脱硫的实验室研究[J].石油炼制与化工,2006,37(1):31-34.
[9]Komintarachat C,Trakarnpruk W.Oxidative desulfurization using polyoxometalates[J].Ind Eng Chem Res,2006,45(6):1853-1856.
[10]赵立明,童仕唐.采用相转移催化氧化法脱除焦化苯中噻吩的研究[J].武汉科技大学学报,2009,32(4):431-435.
[11]佘林源,颜家保,童俊.相转移催化氧化-萃取脱除直馏柴油中硫化物的研究[J].石油炼制与化工,2008,39(7):5-9.
[12]Zhao D,Ren H,Zhao Y,et al.Mechanism and catalytic behavior of quaternary ammonium salts in oxidative desulfurization[J].Petroleum Science and Technology,2009,27(12):1338-1348.
[13]张海燕,杨阳,代跃利.烷基甜菜碱为相转移催化剂的氧化脱硫研究[J].化工科技,2012,20(4):9-11.
[14]赵地顺.相转移催化原理及应用[M].北京:化学工业出版社,2007:144-145.
[15]Huang D,Zhai Z,Lu Y C,et al.Optimization of composition of a directly combined catalyst in dibenzothiophene oxidation for deep desulfurization[J].Ind Eng Chem Res,2007,46(5):1447-1451.
[16]张予辉,叶天旭,孙颖.相转移催化剂催化降低FCC汽油烯烃含量的研究[J].石油学报:石油加工,2008,24(1):34-37.
[17]周二鹏,赵地顺,刘会茹,等.相转移催化氧化脱除噻吩的应用研究[J].化学工程,2008,36(4):34-36.
[18]He L N,Li H M,Zhu W S,et al.Deep oxidative desulfurization of fuels using peroxophosphomolybdate catalysts in ionic liquids[J].Ind Eng Chem Res,2008,47(18):6890-6895.
[19]邱江华,王光辉,曾丹林,等.磷钼酸季铵盐催化柴油氧化脱硫研究[J].武汉科技大学学报:自然科学版,2009,32(4):427-430.
[20]刘帅,冯丽娟,赵玉艳,等.双亲催化剂[(C16H33)N+(CH3)3]3[PMo12O40]-在FCC柴油氧化-萃取脱除含硫化合物中的应用[J].石油与天然气化工,2009,38(2):118-124.
[21]徐亚荣,沈本贤,徐新良,等.磷钨酸/H2O2对富芳汽油氧化萃取脱硫性能的研究[J].化工科技,2009,17(6):12-16.
[22]蔡永宏,贺建勋,邹煜,等.杂多酸季铵盐催化氧化脱除模拟汽油中苯并噻吩[J].西北大学学报:自然科学版,2011,41(4):628-632.
[23]Zhang H X,Gao J J,Meng H,et al.Catalytic oxidative desulfurization of fuel by H2O2in situ produced via oxidation of 2-propanol[J].Ind Eng Chem Res,2012,51(13):4868-4874.
[24]孙智敏.离子液体在燃料油催化氧化脱硫中的应用研究[D].天津:天津大学,2009.
[25]Zhang W,Xu K,Zhang Q,et al.Oxidative desulfurization of dibenzothiophene catalyzed by ionic liquid[BMIm]HSO4[J].Ind Eng Chem Res,2010,49(22):11760-11763.
[26]曹群,陈海丽,赵荣祥,等.乙酸型离子液相转移催化剂氧化脱硫研究[J].当代化工,2010,39(3):245-247,251.
[27]Kedra-Krolik K,Mutelet F,Moïse J-C,et al.Deep fuels desulfurization and denitrogenation using 1-butyl-3-methylimidazolium trifluoromethanesulfonate[J].Energy Fuels,2011,25(4):1559-1565.
[28]Cheng S S,Yen T F.Use of ionic liquids as phase-transfer catalysis for deep oxygenative desulfurization[J].Energy Fuels,2008,22(2):1400-1401.
[29]赵地顺,刘翠微,马四国.FCC汽油光催化氧化脱硫的实验室研究[J].石油炼制与化工,2006,37(6):23-26.
[30]Shiraishi Y,Taki Y,Hirai T,et al.A deep desulfurization process for light oil by photosensitized oxidation using a triplet photosensitizer and hydrogen peroxide in an oil/water twophase liquid-liquid extraction system[J].Ind Eng Chem Res,1999,38(4):1589-1595.
