基于4-(4-咪唑基-亚甲基氨基)苯甲酸配体配合物的合成及结构
2013-08-20陈敏东
徐 静 陈敏东
(江苏省大气环境监测与污染控制高技术研究重点实验室,南京信息工程大学环境科学与技术学院,南京 210044)
Nowadays, crystal engineering is a rapidly expanding field because of its wide application to material and solid state sciences[1-3]. The major goal of this research field is to understand and exploit the potential of weak intermolecular interactions for the construction of novel complex assemblies. While hydrogen bonding is a master key in the area of crystal engineering and the application of intermolecular hydrogen bonds is a well known and efficient tool to regulate the molecular arrangements in crystal structures[4-8]. Meanwhile, the design and construction of metal-organic frameworks (MOFs) are of great interests not only because of their potential applications as functional materials (magnetism,electric conductivity, catalysis and non-linear optics)but also due to their intriguing structural topologies[9-14]. It is known that the nitrogen donor containing carboxylate ligands are useful building blocks for constructing MOFs, and have been utilized to generate zero-, one- (1D), two- (2D), and threedimensional (3D) coordination architectures[15-20].Herein, we designed and prepared 4-[(1H-imidazol-4-yl) methylamino]benzoic acid (H2L) as a logical ligand to construct novel coordination frameworks. The H2L is designed as a multifunctional ligand based on the following considerations: (1) complete or partial deprotonation of H2L can produce L2-or HL-, which are useful ligands for the construction of complexes with novel structures and topologies; (2) the flexible arms of H2L can adopt varied conformations and thus can lead to the formation of diverse structures as well as the complex isomerism which is interesting in crystal engineering. In this paper, we report the complexes [M(HL)2(H2O)2](M (Ⅱ)=Mn (Ⅱ)(1), Cd (Ⅱ)(2),Cu(Ⅱ)(3)) based on the hydrogen bonding interactions.
1 Experimental
1.1 Materials and instrumentation
All the reagents and solvents were used as commercial sources without further purification. FTIR spectra were recorded on a Bruker Vector22 FTIR spectrophotometer using KBr disks. Elemental analyses were taken on a Perkin-Elmer 240C elemental analyzer. Thermogravimetric analyses(TGA) were performed on a TGA V5.1A Dupont 2100 instrument heating from room temperature to 700 ℃under N2with a heating rate of 20 ℃·min-1. The luminescent spectra for the powdered samples were recorded at room temperature on an Aminco Bowman Series 2 spectrophotometer with xenon arc lamp as the light source. In the measurements of the emission and excitation spectra, the pass width was 5.0 nm.
1.2 Synthesis of ligand H2L
This compound was prepared by reaction of paminobenzoic acid with imidazole-4-carboxaldehyde and sodium borohydride. IR (KBr, cm-1): 3 442 (s),1 611 (s), 1 589 (s), 1 538 (s), 1 518 (s), 1 476 (ms),1 380 (s), 1 176 (ms), 844 (m), 788 (s), 641 (ms).
1.3 Synthesis of [Mn(HL)2(H2O)2] (1)
A mixture of MnCl2·2H2O (0.016 g, 0.10 mmol),H2L(0.022 g,0.10 mmol)and H2O(10 mL) were sealed in a 15 mL capacity Teflon-lined reaction vessel and heated at 80 ℃for 3 d. After the reaction mixture was cooled to room temperature, colorless platelet crystals were collected by filtration, washed by ethanol and airdried, yield based on Mn (Ⅱ): 39%. Anal. Calcd. for C22H24N6O6Mn (%):C,50.48;H,4.62;N,16.06.Found(%):C,50.44;H,4.66;N,16.07.IR (KBr pellet,cm-1):3 220 (s), 1 604 (s), 1 553 (s), 1 509 (ms), 1 495 (s), 1 378 (s), 1 242 (s), 1209 (s), 1 188 (s), 1 063 (ms), 850(s),794(s),639(s).
1.4 Synthesis of[Cd(HL)2(H2O)2](2)and[Cu(HL)2(H2O)2](3)
A buffer layer of 4 mL of acetonitrile/water (1/1,V/V) mixed solvent was carefully layered over 4 mL of an aqueous solution of H2L (0.011 g, 0.05 mmol) and NaOH (0.002 g, 0.05 mmol), then 4 mL of an acetonitrile solution of M(NO3)2·6H2O (0.05 mmol,0.017 g for 2; 0.015 g for 3) and was layered over the buffer layer. After two weeks, colorless block crystals of 2 were collected in a yield of 46% based on Cd for 2; brown platelet crystals of 3 were obtained in a yield of 44% based on Cu for 3. Anal. Calcd. for C22H24N6O6Cd (2) (%): C, 45.49; H, 4.16; N, 14.47.Found (%): C, 45.44; H, 4.14; N, 14.49. IR (KBr pellet, cm-1): 3 219 (s), 1609 (s), 1 555 (s), 1 511(ms), 1 495 (s), 1382 (s), 1 244 (s), 1210 (s), 1188 (s),1013 (s), 847 (s), 790 (s), 640 (s). Anal. Calcd. for C22H24N6O6Cu (3) (%): C, 49.67; H, 4.55; N, 15.80.Found (%): C, 49.64; H, 4.56; N, 15.87. IR (KBr pellet, cm-1): 3 221 (s), 1 606 (s), 1 555 (s), 1 510(ms), 1 495 (s), 1 380 (s), 1 244 (s), 1 208 (s), 1 188(s), 1 062 (ms), 849 (s), 791 (s), 638 (s).
1.5 Complex 3 modified glassy carbon (GC)electrode
The GC electrode was first carefully polished with alumina on polishing paper and washed successively with double distilled water and acetone in an ultrasonic bath. About 25 μL of complex 3 suspension (0.25 mg·mL-1) in acetone was cast on the surface of GC electrode and dried in air to form a complex 3 modified electrode (Cu-GCE).
1.6 Crystal structure determination
Crystallographic data of 1~3 were carried out on a Rigaku RAXIS-RAPID Imaging Plate diffractometer at 200 K using graphite-monochromated Mo Kα radiation (λ=0.071 075 nm). The structures of 1~3 were solved by direct methods with SIR92[21]and expanded using Fourier techniques[22-23]. All the nonhydrogen atoms were refined anisotropically on F2by full-matrix least-squares methods. All the hydrogen atoms were generated geometrically and refined isotropically using the riding model. Details of the crystal parameters, data collection and refinements are summarized in Table 1, and selected bond lengths and angles are listed in Table 2. The hydrogen bonding data are summarized in Table 3.
CCDC: 945340, 1; 945338, 2; 945339, 3.

Table 1 Crystallographic data for 1~3

Table 2 Selected bond lengths (nm) and angles (°) for 1~3

Continued Table 2

Table 3 Details of intermolecular hydrogen bonds in 1~3
2 Results and discussion
2.1 Structure description
The reactions of H2L with metal salts lead to the formation of complexes 1 ~3. It is noteworthy that the deprotonation of the carboxylic groups of the H2L ligand in complexes 1~3 was confirmed by IR spectral data,since no IR bands in the range of 1 760~1 680 cm-1were observed in the IR spectra of 1 ~3 (see experimental section), although no base was added in the reaction of complex 1.The band at about 3 433 cm-1is assigned to the ν(O-H)of the water molecules.The IR data are coincident with the crystallographic structural analysis. The results of the X-ray diffraction analysis provide the direct evidence for the structure of the complexes, the same space group and the similar cell parameters of complexes 1 ~3 as listed in Table 1 indicate that the three complexes are isomorphous and isostructual. Thus only the structure of complex 1 is taken as an example to describe here in detail. The complex 1 crystallizes in triclinic space group, the metal center Mn (Ⅱ) ion is six-coordinated by four nitrogen atoms from two different HL-ligands and two oxygen atoms of coordinated water molecules. It is noteworthy that the two oxygen atoms of the deprotonated carboxylic group do not coordinate with the Mn(Ⅱ)atom. When adding excess metal salts in the experimental conditions, no single crystals were obtained from the reactions of HL and Mn (Ⅱ)salts without addition of NaOH as well as from the reactions of HL and Cd (Ⅱ)salts (Cu (Ⅱ)salts) with addition of NaOH.
As shown in Fig.1, the coordination geometry of the Mn (Ⅱ)center can be described as a distorted octahedron with Mn-N bond distances of 0.219 6(2)and 0.237 6(2) nm, Mn-O bond distance of 0.218 9(2) nm(Table 2). An interesting feature of the structure is the presence of inter-molecular hydrogen bonding interactions between the water O-H groups and carboxylate oxygen atoms. The hydrogen bonding data are summarized in Table 3. The single molecules are connected by bonding interactions of O(3)-H(12)…O(1) (symmetry code: -1+x, y, z) to obtain a one dimensional (1D) double chain (Fig.2).

Fig.1 Crystal structure of complex 1

Fig.2 1D infinite double chain

Fig.3 2D layer connected by hydrogen bonds

Fig.4 3D structure of 1
The adjacent 1D chains are further connected together by hydrogen bonding interactions of O(3)-H(11)…O(2) (symmetry code: -x, 1-y, -z), N(1)-H(1)…O(1) (symmetry code: 1+x, y, z) and N(12)-H(9)…O(2)(symmetry code: -1 +x, -1 +y, z) to give a twodimensional (2D) layer structure (Fig.3). A threedimensional (3D) structure as exhibited in Fig.4 is generated and is further consolidated by the C-H…O hydrogen bonding interactions. It gives a nice example of supramolecular framework based on non-covalent interactions of hydrogen bonding interactions.
2.2 Thermal property
Thermalgravimetric analyses (TGA) were carried out to examine the thermal stability of complexes by heating from room temperature to 700 ℃ under flowing N2. Owing to the similarity of the composition and structure of 1 ~3, they showed similar thermal stability and only complex 1 was selected to describe its thermal in Fig.5. The TGA curve of 1 shows that the first weight loss of 6.74% (Calc.: 6.88%) between 160 and 220 ℃ corresponds to the loss of two coordinated water molecules. The decomposition of the residue was observed at ca. 250 ℃and does not end until heating to 700 ℃.
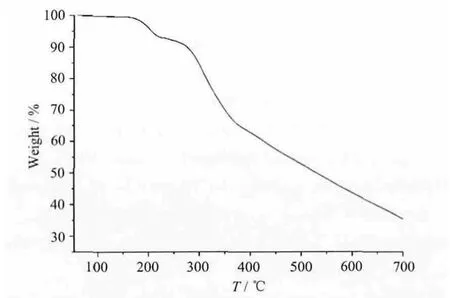
Fig.5 TGA curves of complex 1
2.3 Electrochemical property
The electrochemical analyses usually were conducted under argon with a solution of compound with other electrolyte[24-25].However,compound 3 is insoluble in water and common organic solvents. Thus, the modified glassy carbon electrode (GCE) becomes the optimal choice to study their electrochemical property.The electrochemical study of Cu-GCE was carried out in aqueous phosphate buffer solution(pH=5.5).
Different scan rates (from inner to outer) of 50,100, 150, 200, 250, 300, 350, 400, and 450 mV·s-1.Inset: Cyclic voltammogram of Cu-GCE in pH=5.5 phosphates buffer solution in the potential range of 1.0 to -1.0 V. Scan rate: 100 mV·s-1.
Inset in Fig.6 shows the cyclic voltammogram of 3 in the potential range of +1.0 to -1.0 V, at the modified Cu-GCE, a redox couple attributed to the Cu(Ⅱ)/Cu ガ was observed[26-27], it showed a quasireversible behavior in an aqueous medium[28]. The mean peak potential E1/2=(Epa+Epc)/2 was -33 mV vs.SCE for the Cu-GCE. In fact, there is a sharp oxidation peak due to Cu (Ⅱ)+e-→Cu ガwhile the reduction peak is very broad. This is most possibly due to the instability of the reduced form of the complex with different coordination geometry which can undergo a fast chemical oxidation in an aqueous solution of pH =5.5[26]. Scan rates effect on the electrochemical behavior of the Cu-GCE was investigated in the potential range of +1.0 to -1.0 V in pH =5.5 phosphates buffer aqueous solution, as shown in the Fig.5. When the scan rate was varied from 50 to 450 mV·s-1, the peak potentials change gradually: the cathodic peak potentials shifted to negative direction, namely, the peak-to peak separation between the corresponding cathodic and anodic peaks increased, but the mean peak potentials did not change on the whole.

Fig.6 Cyclic voltammogram of Cu-GCE in pH=5.5 phosphates buffer solution at different scan rates
3 Conclusions
Three novel complexes [M(HL)2(H2O)2](M(Ⅱ)=Mn(Ⅱ)(1), Cd (Ⅱ)(2), Cu (Ⅱ)(3)) were synthesized under different conditions, and the structures are further connected by hydrogen bonds interactions resulting in formation of 3D structures. The hydrogen bonding interactions are the most prominent of supramolecular interactions, not only in synthetic systems but also in nature. In addition, the thermal stability property of 1 and electrochemistry property of 3 are investigated.The results showed that the metal centers have remarkable influence on the property of the complexes. This work provides useful information for employing carboxylate and imidazolate/imidazolecontaining ligand to assemble functional coordination polymers with novel structure and property.
[1] Bernstein J, Davey R J, Henck J O. Angew. Chem. Int. Ed.,1999,38:3441-3461
[2] Eckert H, Ward M. Chem. Mater., 2001,13:3061-3083
[3] Rao C N R, Natarajan S, Vaidhyanathan R. Angew. Chem.Int. Ed., 2004,43:1466-1496
[4] Helay F, Maris T, Wuest J D. Cryst. Growth Des., 2008,8:4517-4525
[5] Manzano B R, Jalón F A, Soriano M L, et al. Cryst. Growth Des., 2008,8:15851594
[6] Li Y P, Yang P. Chin. J. Chem., 2007,5:1715-1724
[7] Yang X G, Li D S, Fu F, et al. Chin. J. Chem., 2008,26:655-660
[8] ZHANG Li(张丽), NIU Su-Yun(牛淑云), JIN Jin(金晶),et al. Acta Chim. Sin.(Huaxue Xuebao), 2007,65:1032-1038
[9] Cote A P, Benin A I, Ockwig N W, et al. Science, 2005,310:1166-1170.
[10]Yaghi O M, O′Keeffe M, Ockwig N W, et al. Nature, 2003,423:705-714
[11]Wu C D, Hu A, Zhang L, et al. J. Am. Chem. Soc., 2005,127:8940-8941
[12]Fang Q R, Zhu G S, Xue M, et al. Cryst. Growth Des., 2008,8:319-329
[13]Koh K, Wong-Foy A G, Matzger A J. Angew. Chem. Int.Ed., 2008,47:677-680.
[14]Song Y, Zhang P, Ren X M, et al. J. Am. Chem. Soc., 2005,127:3708-3709
[15]Yigit M V, Biyikli K, Moulton B, et al. Cryst. Growth Des.,2006,6:63-69
[16]Gao H L, Ding B, Yi L, et al. Inorg. Chem. Commun., 2005,8:151-153
[17]Gao H L, Yi L, Ding B, et al. Inorg. Chem., 2006,45:481-483
[18]Ghosh S K, Bharadwaj P K. Eur. J. Inorg. Chem., 2005,4886-4889
[19]Wang H S, Zhao B, Zhai B, et al. Cryst. Growth Des.,2007,7:1851-1857
[20]Koh C K. Chem. Soc. Rev., 2002,31:157-167
[21]SAINT, Pragram for Data Extraction and Reduction, Bruker AXS, Inc.; Madison, WI, 2001.
[22]Sheldrick G M. SHELXS-97, Program for Crystal Structure Solution, University of Göttingen, Germany, 1997.
[23]Sheldrick G M. SHELXL-97, Program for Crystal Structure Refinement, University of Göttingen, Germany, 1997.
[24]Hellyer R M, Larsen D S, Brooker S. Eur. J. Inorg. Chem.,2009,1162-1171
[25]Nicola C D, Garau F, Karabach Y Y, et al. Eur. J. Inorg.Chem., 2009,666-676
[26]Wang X L, Lin H Y, Liu G C, et al. J. Organomet. Chem.,2008,693:2767-2774
[27]Wang X L, Zhao H Y, Lin H Y, et al. Electroanalysis,2008,20:1055-1059
[28]Younathan J N, Wood K S, Meyer T J. Inorg. Chem.,1992,31:3280-3285
猜你喜欢
杂志排行

无机化学学报的其它文章
- Synthesis,Crystal Structure and Antibacterial Activity of 2D Hydrogen-bonds Layered Magnesium(Ⅱ)Complex
- The Uptake and Membrane Transport of Cesium in Human Erythrocytes
- Preparation of Functionalized Graphene Sheets via Microwave-Assisted Solid-State Process and Their Electrochemical Capacitive Behaviors
- A New 2D Layer Manganese(Ⅱ)Complex Assembled by Flexible 1,4-Benzenebis(thioacetic acid)Ligand:Synthesis,Crystal Structure and Magnetic Property
- Synthesis of a New Cu(Ⅱ)Complex with in situ Generated 3-Hydroxy-2,4,6-pyridinetricarboxylate Ligand and Analysis of the Reaction Mechanism
- A New Nickel(Ⅱ)Complex Based on Novel Pyridyl-carboxylate Schiff-Base Ligand:Synthesis and Crystal Structure