美国转基因食品自愿咨询程序初论
——基于食品药品监督管理局指导政策的分析
2013-08-11刘旭霞
刘旭霞 刘 钰
美国转基因食品自愿咨询程序初论
——基于食品药品监督管理局指导政策的分析
刘旭霞 刘 钰
为应对“转基因管理的协调框架”的要求,美国食品药品监督管理局建议企业在转基因食品研发之初和上市之前对转基因食品和蛋白质的安全性问题进行咨询,这就是转基因食品自愿咨询程序。可以说,转基因食品自愿咨询程序是该机构对转基因食品管理的主要方式。分析转基因食品自愿咨询程序的演进过程,厘清转基因食品自愿咨询程序中的管理机构、具体流程和实质要件,可以发现美国食品药品监督管理局对转基因食品的基本态度和咨询程序的内在机理。
转基因食品;自愿咨询程序;食品药品监督管理局;指导政策;实质等同原则
刘旭霞,华中农业大学文法学院教授。(湖北武汉 430070)
刘 钰,苏州市中级人民法院书记员。(江苏苏州 215007)
美国白宫科技政策办公室(OSTP)制定的“转基因管理协调框架”(以下简称管理协调框架)要求转基因生物(GMO)应当在现有法律框架下进行授权和管理,[1]并对食品药品监督管理局(FDA)、环境保护部(EPA)以及农业部(USDA)在转基因管理方面的权限和职责进行了划分,其中FDA管理的对象是转基因食品(GMF)。基于“实质等同”原则,FDA采取“转基因食品自愿咨询程序”这一宽松形式对转基因食品予以监管。所谓自愿咨询程序(以下简称 “咨询程序”)是指:开发商在进行 GMF生产研发、安全评估期间以及上市之前,主动对GMF及其新蛋白的安全性问题向FDA咨询,最终完成食品安全评估并将咨询内容以特殊文件形式公开的程序。依据阶段的不同,咨询程序又分为前期咨询程序 (Initial Consultations)和最终咨询程序(Final Consultations)。在 GMF研发的早期,开发商就可以针对GMF在安全评估中的问题咨询FDA,这一阶段被称为前期咨询程序。在经过反复咨询之后,由FDA最终确定GMF的安全性,此时开发商需要制作证明 GMF安全性和咨询全过程的文件并公开,这一阶段被称为最终咨询程序。虽然咨询程序强调“自愿”,但由于FDA咨询的权威性,开发商为了赢得消费者的认可,大都自发选择了这种“自愿”程序。[2]
一、转基因食品自愿咨询程序的制度演进
从1992年开始,FDA相继颁布了五份指导性文件来帮助开发商进行食品安全评估并完成咨询程序:《源于转基因食品的食品政策——转基因食品工业指南》初步形成了自愿咨询程序的框架结构,《关于源于植物新品种食品的咨询程序政策》将咨询程序分为前期咨询和最终咨询两部分,《使用抗生素抗性标记基因的转基因植物中的指南草案》和 《食用新植物非杀虫蛋白的食品安全评估建议》针对两类特殊转基因食品的自愿咨询作出了规定,《生物技术最终咨询程序—生产者向食品添加剂安全办公室提供电子或纸张申报书的指导》则给开发商提供一份最终咨询文件的模板。
早在20世纪80年代末,公众就开始针对GMF问题向FDA咨询。1992年,FDA颁布了指导性文件《源于转基因食品的食品政策——转基因食品工业指南》[3](以下简称 《工业指南》),该指南一开始便指明了FDA对GMF的基本态度并明确了对遗传修改技术、DNA重组技术的支持。此外,《工业指南》还针对GMF所产生的效果、营养、新蛋白、过敏原性等问题做出了回应。在《工业指南》的最后部分,FDA向开发商提供了一个食品科学评估的依据,来判断新的转基因植物品种是否和受体品种一样安全。
1992年《工业指南》指出,新品种开发人员尽可能就GMF的安全和监管问题进行咨询。随着转基因技术发展,开发商开始针对进入市场的具体程序致函FDA。此外,因隶属于FDA的食品安全应用营养中心(CFSAN)的上市审批办公室(OPA)和兽药中心(CVM)的重组,导致《工业指南》的部分咨询程序需要调整。为了应对变化,1996年,OPA/CFSAN与CVM的合规管理办公室(OSC)联合颁布了 《关于源于植物新品种食品的咨询程序政策》[4](以下简称《咨询政策》),这项指导意见主要是对《工业指南》最后一部分咨询程序的细化,包括开发商向机构咨询的方法及CFSAN、CVM处理这些咨询的程序等。《咨询政策》针对来源于植物新品种食品①的咨询过程和咨询程序作出规定,第一次明确了 “前期咨询的程序”以及“最终咨询程序”的地位、作用和流程,并划分了FDA内部各机构的分工及“生物技术评估委员会”(BET)的职能,这是整个咨询程序的关键和核心内容。
FDA于1998年公布了 《使用抗生素抗性标记基因的转基因植物中的指南草案》[5](以下简称《抗性基因指南》)。在正文部分,《抗性基因指南》主要针对需要咨询的内容、咨询形式和专家名单、咨询摘要等问题做了简要概述,明确了可用于开发的标记基因的植物品种的咨询。
依据OSTP的规定,USDA负责监督GMO的实地测试。然而,由于从试验到商业化过程中的花粉漂移以及种子混合可能会导致食品中转基因物质的低水平呈现,所以,即使是那些已经得到USDA批准的GMO也存在一定风险。基于此,2006年,CFSAN与CVM又共同颁布了《食用新植物非杀虫蛋白的食品安全评估建议》[6](以下简称 《评估建议》)。主要解决非杀虫蛋白的评估及前期咨询问题。此外,该建议还具体介绍了新蛋白质的早期评估所应考虑的主要内容、食品安全早期评估所应递交的材料内容和格式、食品安全早期评估的具体流程等问题。总体来讲,《评估建议》是对《工业指南》和《咨询政策》中的前期咨询程序的补充和解释,在内容上有一定的重合。值得说明的是,《评估建议》仅仅适用于非杀虫蛋白的前期咨询,对于杀虫蛋白,则须依照 《工业指南》和《咨询政策》进行咨询。
为了给开发商提供一份最终咨询文件的模式范本,FDA于2010年颁布了 《生物技术最终咨询程序—生产者向食品添加剂安全办公室提供电子或纸张申报书的指导》[7](以下简称《最终指导》),《最终指导》规定了生物科技最后咨询意见的一般信息、一份电子形式的新的蛋白质咨询意见。开发商在最终咨询完成之后,将依据《最终指导》制作相应的电子文档并将文档递交到食品添加剂安全办公室(OFAS),最终完成咨询程序。
从1992年FDA颁布第一项指导文件开始,FDA始终对GMF持一种较为宽松的态度。但是,随着基因技术的发展和应用,开发商开始遇到更多新的问题需要咨询,公众也开始提出更多新的质疑。因为技术的进步和指导文件本身内容的缺陷,后来FDA又颁布了新的指导,一方面起到了制度补充的作用,解决了旧问题;另一方面建立了新的制度,解决了新问题。从上述分析看,上述五个指南共同构成了FDA对 GMF管理的法规框架(详见表 1)。
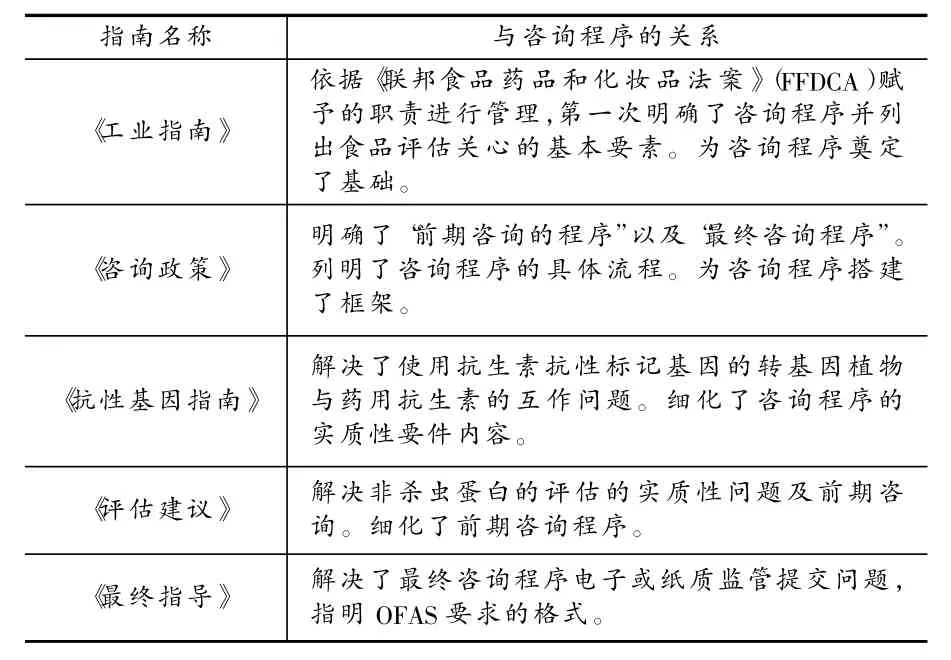
表1 FDA各指导文件及其联系
二、FDA在转基因食品自愿咨询程序中坚持的基本原则
转基因食品自愿咨询程序基本原则包括实质等同、分类管理、协调管理三个层面。
(一)实质等同原则
“实质等同”指假如转基因产品在营养成分、用途、过敏源等方面与传统产品不存在区别,则只需以同于传统产品的模式进行管理,不需要增加特殊的检测、审批,不需要强制标签,只有存在可靠的科学证据证明,该类转基因产品存在一定风险并且有可能造成损害时,政府或监管机构才能采取管制措施的基本监管原则。在1992年的《工业指南》中,FDA指出:“在大多数情况下,由于植物基因改造而成为食物的物质构成会与蛋白质、脂肪、油脂和碳水化合物等物质一样或类似。”从FDA的阐述中可以看出,GMF的评估理念是“结果主义”,即以产品为基础(Product-based)的管理模式,监控管理的对象是生物技术产品,而不是生物技术本身。FDA仅关注于其最终的食品营养特性和是否产生毒理作用,也就是说,FDA推定GMF与一般食品“实质等同”。
(二)分类管理原则
1.依照GRAS管理转基因食品中的残留蛋白
GRAS是Generally Recognized As Safe(公认安全)的缩写,是美国食品法律中一个非常重要、独特和庞杂的食物成分类别。在美国食品法律和法规的位阶上,GRAS介于常规食物成分和食品添加剂之间。[8]1958年,为了回应民众对食品制造过程中化学品增加的质疑,国会依据FFDCA(《联邦食品药品化妆品法》颁布了 《食品添加剂修正案》(以下简称《修正案》)。与其他法律相比,《修正案》为食品添加剂建立了一个上市前批准程序,其中还规定生产商必须要向 FDA证明这种添加剂的安全性。如果食品添加剂没有获得许可,在法律上就是不安全的。同时《修正案》根据GRAS制度排除了一些应当管制的食品添加剂:一种是一些源于自然界的食品添加剂,另一种是某些化学物质添加剂。这两类GRAS添加剂在没有经过FDA的正式审查下,也可以合法上市销售。当然,关于何种添加剂属于GRAS,不能简单地依据上述的标准进行判断。对此,《工业指南》指出,FDA鼓励新食品成分的生产商向FDA咨询当新成分是否是GRAS,比照FFDCA对食品添加剂的管理建立了一套针对GMF残留蛋白的管理制度。
2.抗生素抗性标记转基因食品的特殊处理
FDA认为从植物中抗生素抗性标记转移到在肠道或的可能性不大。在《抗性基因指南》中,FDA指出,如果抗生素抗性表现在食物中,评估应包括一个由基因编码的蛋白质或酶的安全性进行评估。《抗性基因指南》提出了建议参考的因素,包括:(1)评估潜在毒性的蛋白质;(2)评估是否有可能引起过敏反应的蛋白质;(3)评估抗生素抗性标记基因所编码的食品中的酶或蛋白是否会损害口服抗生素治疗效果。
(三)协调管理原则
美国以“实质等同原则”为基石,认为目前还没有证据证明转基因产品会对人体健康或生态环境产生危害,因此,转基因技术及其产品是安全的。[9]基于此,美国建立的管理协调框架,分别将种植安全、食用安全及农药豁免交由USDA、FDA以及EPA进行管理,这就使得FDA在咨询程序中要充分考虑与相关制度的协调。
1.针对转基因食品安全评估与USDA的协调
《特定转基因生物的进口、洲际转移和环境释放》[10]中,在USDA关注转基因作物是否派生为植物劣性杂草或病虫害的同时也对转基因蛋白的管理进行了规定,比如USDA将 “该转基因生物对其他生物体造成损害或者伤害的毒性、过敏性以及能力”这一标准作为申请解除管制程序(deregulation)的考量之一。也就是说,开发商在进行自愿咨询前,如果拟上市的GMF所对应的(来源)转基因作物已经得到解除管制,则转基因作物的食用安全性已经得到了证明,由此也可以推断GMF的安全性。所以,FDA采取咨询程序在制度层面的原因是USDA对转基因作物食用安全性的监管。
2.针对抗虫类转基因食品与EPA的协调
当植物具有抗虫转基因(植物内置式农药)的时候,EPA主要负责转基因植物销售与推广前内置式农药的登记,并规定了其食品中的残留量的最低容忍及免除条件。也就是说,抗虫性GMF的监管权在协调框架下出现了管理的重叠。因此,FDA在《工业指南》中指出FDA和EPA就《联邦杀虫剂、杀菌剂和灭鼠剂法》(FIFRA)定义的杀虫剂受EPA的管制这一问题达成一致。对于与杀虫剂有关的食品安全问题,包括用来证明简要基因存在的标志基因,应当由EPA来管辖。除了和杀虫剂有关的问题,任何其他的安全问题都在FDA的管辖范围内,并且应该根据《工业指南》来进行管理。
三、转基因食品自愿咨询程序的制度安排
具体实施指导评估和咨询的机构是BET。依据 《咨询政策》OPA/CFSAN和OSC/CVM设立BET,指明监管和需要处理的科学问题。一旦所有相关问题已经得到充分的解决,将结束咨询程序。BET的核心团队一般由消费者安全机构官员(CSO)、分子生物学家、化学家、环境科学家、毒理学家及营养学家组成。值得说明的是,FDA并不直接参与食品安全评估,而是建议开发商在评估过程中能尽可能多地咨询BET,以确定食品安全评估的准确性及材料的完整性。在《工业指南》中,FDA列出了开发商在研发和评估食品中需要注意的若干问题,如意想不到的效果、已知的有毒物质、营养、新物质、变应 (过敏)原性等,具体程序如下。
(一)前期咨询程序
“前期咨询程序”简称 “前期程序”。在 《工业指南》中,FDA建议开发商针对植物新品种进行咨询,讨论其进入市场之前相应的科学和规制问题。《咨询政策》中FDA继续鼓励开发商在开发阶段早期咨询他们的产品,此类咨询将有助于解决安全、营养、和法规问题。其中,针对非杀虫蛋白的,FDA还规定了新型蛋白咨询(NPC),具体指开发商在早期食品安全评估期间,提前就一种由新的作物品种产生的、计划被用做食品的蛋白通知FDA。
在《工业指南》中,FDA并没有直接提出需要提交何种材料来申请,而是以建议的形式来帮助开发商完成食品安全评估并递交申报的材料。针对新蛋白的评估,《工业指南》第七部分F指出,根据情况,被引入的蛋白质的安全评估应该包括已知或被怀疑的致敏性;加工的影响;已知或潜在的毒性等几个方面。针对评估的具体要求和材料的细节,《咨询政策》做出了回应,其中指出,FDA鼓励开发商依据国际食品法典委员会 《重组DNA植物食品安全评估准则》[11]的要求进行。
《咨询政策》和 《评估建议》指明了前期程序的具体流程,此外咨询还需受CFSAN的调整。具体来讲,开发商提交咨询材料,FDA收到材料后,将咨询请求送达BET进行协调咨询,请求将被登记在案。之后,BET的主席会完成一个“联系表单”,并连同咨询请求一同转到文件控制中心 (DCC),最终建立一个生物技术通知文件(BNF)。同时这些材料的副本将送达CFSAN的生物技术战略管理处。DCC收到表单后,建立BNF并指定BNF号码,并进入到科学信息检索和交换网络(SIREN)。主席在收到材料后15个工作日内回应开发商并确定咨询见面会的地点和日期。此时,在收到材料的120个工作日内,由BET进行评估。在这之后,主席会和开发商进行一次关于科学、监督和调控问题等方面的咨询会议。同时,BET将组成一个团队对相应的议题进行评估并提出建议。最后,由FDA公开数据,完成初步评估,确定蛋白质是否存在食品安全问题。
(二)最终咨询程序
“最终咨询程序”简称 “最终程序”。《咨询政策》规定,在研究开发过程的每个阶段,开发商都应该积累充足资料以确保其产品是安全的。按照FFDCA的规定,在这期间开发商还必须持续不断地向FDA进行咨询。在咨询程序的最后阶段,由FDA建议开发商提交安全和营养评估总结,以最终确定和完成整个咨询程序,而这个过程,就称为“最终咨询程序”。开发商在最后,将形成一份“FDA最终咨询文件”(PBN)。PBN文件作为GMF上市前的官方文件,在食用安全性上有较高的证明力和权威性。依据《咨询政策》的规定,最终咨询文件所包含的信息包括:关于通过介绍遗传物质编码表示产品特性和功能的信息,对任何有关生物工程作物和食品产品衍生品表达的汇总评估;关于产品已知的或可疑的过敏性及毒性阐述以及其他任何关于生物工程食品安全性和营养性的评估信息。
当开发商积累了足够的信息能够证明产品已经符合FFDCA及FDA指导的时候,开发商则可以提交安全和营养评估的总结,以启动最终咨询程序,完成GMF上市前的最后程序。其具体的流程如下,主席收到材料后在收到申请后的10天内,将复印的总结分发给BET成员进行评估。在收到材料后4周内,主席会从BET成员处得到反馈信息,以判断开发商在总结中提到的安全或监督信息是否充足。基于BET的建议,主席将反馈开发商拟咨询会结果的草稿并最终由CFSAN的负责人签字并公布。
四、美国转基因食品自愿咨询程序对我国的启示
FDA针对GMF采取相对宽松的咨询程序,有利于美国转基因技术的研发和推广,其背后包含了诸多经济、文化和制度因素,如美国是世界上转基因作物研发最早的国家,转基因产品的商业化程度一直处于世界领先地位等。[12]自1985年第1批抗病毒、抗虫害的转基因植物进入田间试验以来,转基因安全审批的数量逐年提高,直至1996年,转基因作物开始大量商业化种植。[13]为了维持转基因产业化水平和市场占有率,在世界范围内推广转基因技术及产品,美国采用咨询程序可以大幅度提高审批效率。此外,在美国开发商为研发主体,这些开发商从研发初期便投入了巨大的资金,基于利益导向,开发商非常注重与大众的沟通,同时与FDA磋商,经过不断的制度修正,最终形成现行的咨询程序。[14](P94-98)在事后救济制度方面,美国采用产品质量的严格责任制度,一旦符合严格责任,开发商将承担巨额惩罚性赔偿,[15]为避免因赔偿带来的损失,开发商一般都会主动咨询。虽然依据我国目前的现实国情还不具备采用自愿咨询程序的条件,但其中仍有的诸多经验值得我国转基因生物生产应用安全审批制度去借鉴。
(一)充分保障公众参与
在美国,FDA任何一部指导性法规的颁行都是为了回应当时公众和科学家对GMF提出的新问题,如1994年前后,一些转基因开发商针对他们进入市场的计划致函FDA,《咨询政策》即随之应运而生。此外,FDA通过咨询程序,建立起了良好的“开发商—机构—公众—专家”沟通交流和互动机制。咨询程序的制度设计是在开发商的咨询、公众的质疑、专家的参与等多因素共同推动下完成的。《最终指导》还指出,咨询内容还应当制作一份档案,以备公众查阅。纵观我国有关转基因方面的安全立法,都是通过审批、申报等规则来管理的,明显缺乏现代社会立法应当采用的激励机制。预防转基因技术带来的健康问题和生态风险,不仅是国家的职能,同时也是公民的权利和责任。“政府包揽一切”的做法会导致社会参与的缺位,导致我国转基因作物产品安全不能得到全面和深入的监管。[16](P78)对此,我国首先应当做到信息公开,特别是涉及安全评价的相关数据和过程公开。此外,评价机构应当及时的回应公众的质疑,有必要时还应对不完备的部门规章予以修订。通过公众参与,进一步形成良好的“开发商—机构—公众—专家”沟通机制。
(二)合理划分审批机构的权力范围
美国设置的管理协调框架实际上是对原有制度的整合、派生,EPA、USDA、FDA的管理权限也都是参照传统的作物、食品甚至农药。具体到FDA,其将GMF中的部分转基因蛋白视为常规食品中的食品添加剂并参照GRAS进行管理的方式,在本质上仍然是原有制度体系的派生,对 GRAS的豁免同样的应用到了转基因蛋白上。此外,FDA并没有设置新的机构来专门回应咨询问题,其主管机构仍然是其下属的CFSAN和CVM。可见,美国转基因安全管理合理划分了权力范围,也相应简化了审批的流程。反观我国,在实际操作中,由于《新资源食品管理办法》过于粗略,缺乏可操作性,而农业部颁布的 《农业转基因生物安全评价管理办法》针对食品安全评估中的细节做了详细的规定,使卫生部与农业部职能出现重叠。对此,应当理顺农业部与卫生部在GMF安全评价中的关系。对转基因作物(种子)、GMF采用不同的方式区别管理。卫生部应当效仿FDA,有效利用现有的制度体系,理清与农业部的关系。具体来说,可以由农业部负责安全评价和审批,而卫生部负责具体转基因生物加工产品的管理。与此同时,在食品安全评价方面,可以借鉴和引入国际食品法典委员会的标准。
(三)适度减轻开发商负担
咨询程序减少了烦琐的审批,减轻了开发商的时间和成本压力。虽然开发商为了赢得消费者的认可,大都自发选择了咨询程序,使得本为“自愿”的程序具有了无形的 “强制性”,但相对于其他国家和地区,这种管理方式依然具有较强的灵活性,在一定程度上减轻了开发商的负担。如在《工业指南》中,FDA建议开发商讨论在进入市场之前其科学和规制问题,其中开发商可以自愿选择咨询的方式、内容甚至次数,而FDA也可以依据实际情况确定BET的具体组成。针对需要提交材料的内容,FDA也仅仅是以建议的形式来发布可供参考的样本。FDA并没有设置新的机构来专门回应咨询问题,其主管机构仍然是其下属的CFSAN和CVM。对此,我国卫生部借鉴美国经验,针对食品安全评价做出全过程的指导,针对农业部已经予以评价的审批事项适度简化了程序。这不但可以充分节省开发商的评估成本,还可以压缩从研发到上市的时间。
(四)及时调整现行制度
从1992年确立咨询程序开始,咨询程序就经历了多次修订和完善。FDA任何一部指导性法规的颁行都是为了回应当时公众和科学家对GMF提出的新问题。《工业指南》、 《咨询政策》、 《抗性基因指南》、 《评估建议》、《最终指导》的公布,都与当时转基因技术的发展相适应。对此,我国应当以现行行政法规、部门规章为基础,完善相应安全评估指导和政策性指导。根据转基因食品的安全性以及公众的态度对现有的申请政策予以适度调整,及时回应公众质疑并完善具体审批细节。
经过多年的发展,美国自愿咨询程序已成为促进美国转基因产业快速发展的重要制度之一。FDA在咨询程序中,一方面具有指导安全评价的职能,另一方面也起到了产业引导的作用。我国作为转基因技术的研发大国,应当继续对生物技术产业予以支持,并学习和借鉴美国的自愿咨询程序,为转基因技术的研发和推广提供制度支撑并做好产业规划和指导。在转基因食品的审批程序上,要适度减轻开发商的负担。同时,建立起良好的“开发商—机构—公众—专家”沟通交流和互动机制,做到信息公开,及时回应公众的质疑。只有这样,才能实现我国转基因技术产业的健康、可持续发展。
注释:
①实际上,FDA基于“实质等同原则”,将“转基因食品”(GMF),参照“源于植物新品种食品”(Foods Derived from New Plant Varieties)进行管理。在本质上,各指导文件针对“源于植物新品种食品”的规定也都同样适用于GMF。因此,后文中提到的“源于植物新品种食品”指GMF。为了保证引用文献的准确性,笔者不对“源于植物新品种食品”做过多的限制性解释。
[1]U.S.Congress OTA.New Developments in Biotechnology—Field Testing Engineered Organisms,Genetic and Ecological Issues,no.3,OTA-BA-350,May 1988.Washington, GPO, 1988.p.60.http://www.fas.org/ota/reports/8816.pdf.
[2]刘旭霞.美国转基因管理协调框架下的安全审批制度初论——以制度演进为视角[J].自然辩证法通讯,2012,(5).
[3]FDA:Statement of Policy-Foods Derived from New Plant Varieties- Guidance to Industry for Foods Derived from New PlantVarieties,http://www.fda.gov/Food/Guidance Compliance Regulatory Information/Guidance Documents/Biotechnology/ucm096095.htm.
[4]FDA:Consultation Procedures under FDA's 1992 Statement of Policy- Foods Derived from New Plant Varieties.http://www.fda.gov/Food/Guidance Complian-ce Reg ulatory Information/Guidance Documents/Biotechnology/uc-m096126.htm.
[5]FDA:Guidance for Industry Use of Antibiotic Resistance Marker Genes in Transgenic Plants Draft Guidance.http://www.fda.gov/Food/Guidance Compliance Re-gulatory Information/Guidance Documents/Biotechnology/ucm096135.htm.
[6]FDA:Guidance for Industry:Recommendations for the Early Food Safety Evaluation of New Non-Pesticidal Proteins Produced by New Plant Varieties Intended for Food Use.http://www.fda.gov/Food/Guidance Compliance Regulatory Information/Guidance Documents/Biotechnology/ucm096156.htm.
[7]FDA:Biotechnology Final Consultations Guidance for Industry Providing Regulatory Submissions in Electronic orPaper Format to the Office of Food Additive Safety.http://www.fda.gov/Food/Guidance Compliance Regulatory Information/Guidance Documents/Food Ingredientsand Packaging/Regulatory Submissions/ucm199073.htm.
[8]史晓伟.美国“公认安全使用物质”(GRAS)法规简介[J].中国食品添加剂,2009,(4).
[9]John S Applegate.The Prometheus Principle:Using the Precautionary Principle to Harmonize the Regulation of Genetically Modified Organisms.Indiana journal of Global Legal Studies,2001,Vol.9.
[10]Importation,Interstate Movement and Release Into the Environment of Certain Genetically Engineered Organisms(7 CFR Part 340)Federal Register/Vol.73,No.197/Thursday,October 9,2008/Proposed Rule.shttp://www.regulations.gov/Guidance document Detail;D=APH IS-2008-0023-2583.
[11]联合国粮农组织.重组DNA植物食品安全评估准则 [EB/OL].http://www.fao.org/docrep/008/y5819c/y5819c03.htm.
[12]陈德敏,邓禾.对我国转基因食品安全性的立法探讨[J].重庆大学学报(社会科学版),2004,(3).
[13]刘定富.美国主要作物转基因商业化进展及启示[J].中国种业,2010,(12).
[14]John Davison,GM plants:Science,politics and EC regulations.Plant Science,2010,Vol.178.PP.72-74.
[15]王学军.美国产品责任法中严格责任原则的确立与理性回归[J].广西社会科学,2001,(5).
[16]樊龙江,周雪平.转基因作物安全性争论与事实[M].北京:中国农业出版社,2001.
【责任编辑:胡 炜】
D912.29
A
1004-518X(2013)12-0055-06
国家社科重点项目“转基因农业生物技术产业化可持续发展研究”(11AZD107)、国家重大科技专项重大研究课题“转基因生物新品种培育”(2011ZX08001-001)
