劣质重油脱碳改质接触剂上焦炭气化反应规律
2013-07-19霍金丽徐晓枫高金森
田 刚,王 刚,霍金丽,徐晓枫,高金森
(中国石油大学 重质油国家重点实验室,北京102249)
随着常规石油资源日益减少,储量巨大的非常规石油资源——稠油、超稠油、油砂沥青等劣质重油资源的高效转化利用越来越受到关注[1-3]。劣质重油一般具有H/C原子比低,胶质、沥青质含量高,残炭值高,密度、黏度大,杂原子、金属含量高等特点,采用传统重油加工工艺加工劣质重油存在许多未解决的难题。例如,采用重油催化裂化技术处理劣质重油,催化剂易因重金属中毒而造成不可逆失活[4];固定床加氢技术一般不能用于(Ni+V)质量分数高于150μg/g和残炭质量分数大于15%的原料[5],而劣质重油的重金属含量和残炭值远高于此值;延迟焦化在原料性质变差时遇到的首要问题是加热炉辐射炉管结焦倾向严重,致使装置不能正常操作;另外,采用以焦化为龙头的重油加工工艺所得轻质油收率低,产品质量差,劣质高硫焦产率高[6-7]。针对这些情况,王洪亮等[8]开发了以廉价材料制备的具有适宜微反活性的多孔固体微球接触剂,对劣质渣油进行流化脱碳、脱金属的改质反应,为进一步加工利用劣质重油创造了条件。以脱碳为主要目的之一的接触剂结焦失活后,其含炭质量分数大多在2%以上,利用空气烧焦再生产生的热量将远超过装置自身热平衡的需要。因此,如何控制再生过程热平衡、合理利用过剩热量是一个亟待解决的问题。
近年来,由于原料油变重,导致FCC催化剂上积炭量增加,随之带来的系统热平衡问题已经引起了很多研究者的关注。Mayes[9]提出,利用贫O2与催化剂上积炭燃烧反应生成CO,从而减少放热,调控热平衡,但需要增加CO燃烧器才能实现该目标。此外,一些研究者提出,利用CO2、H2O(g)及其混合气体等与催化剂上焦炭反应,达到除焦再生的目的。Hettinger[10]提出,在催化剂再生过程中,采用CO2作为气化剂与流化剂代替空气,与催化剂上积炭反应生成CO,达到减少放热的目的。但焦炭-CO2在常规再生温度下反应速率很低,因而该设想的实际应用受到限制。吴治国等[11]提出,利用重质油加工过程中的过剩炭,通过H2O(g)与催化剂上焦炭的气化反应生产水煤气(H2+CO),将过剩热量转移到价值较高的气体中,并可有效调控系统热平衡。Corma等[12]讨论了一种新型FCC催化剂上焦炭与 H2O(g)、O2-H2O(g)混合气等反应对系统热平衡的影响,结果表明,再生温度略高于1023 K时,利用水蒸气重整的方式可有效调控再生热平衡,并副产部分价值较高的H2。因此,在常规烧焦再生过程中引入部分焦炭气化过程,将过剩焦炭热值转移至附加值较高的气体中,不失为一个解决高含炭量催化剂再生热平衡问题的较佳途径,同时可减少烧焦再生过程中CO2的排放。
通常条件下,在温度低于1073K时,焦炭与CO2、H2O(g)气化反应速率较慢,导致烧焦强度很低;而高于1073K时,容易造成常规FCC催化剂的不可逆失活。与常规FCC催化剂相比,接触剂可以在较高温度水热环境下再生,并保持活性及孔结构的稳定。笔者采用微型固定床反应器,考察劣质重油脱炭改质接触剂上焦炭与 O2、H2O(g)、O2-H2O(g)混合气等气化剂的再生反应规律,探索不同气化剂条件下,反应温度、气化剂体积分数、反应时间等因素对产物气体组成以及反应速率的影响,旨在为劣质重油流化脱炭接触剂气化再生工艺提供思路。
1 实验部分
1.1 接触剂的性质及结焦的制备
实验所用接触剂由中国石油大学重质油国家重点实验室采用喷雾成型的方法制备,其基本性质列于表1。使用前,在水热老化装置上用100%水蒸气在1173K下进行流化老化处理8h。以密度0.9938g/cm3(293K 时)、残炭值17.82%、金属总质量分数275.1μg/g的辽河减压渣油为原料油,在小型固定流化床上反应,接触剂积炭后可得到含炭质量分数为2.45%的待生剂。

表1 本实验所用接触剂的性质Table 1 Properties of catalyst for the experiment
1.2 微型固定床气化装置及流程
图1为微型固定床气化装置示意图。

图1 待生接触剂上焦炭气化用微型固定床气化装置示意图Fig.1 Schematic diagram of micro-fixed bed gasification setup for coke on spent catalyst
该装置由进气系统、温度控制系统、反应系统和产物分离收集系统4部分组成。反应器内径10mm、长370mm,将1g待生接触剂置于反应器中部石英棉床层上,在N2保护下,以20K/min速率升至反应所需温度。待温度稳定后,切换反应气体进入反应器与接触剂上的焦炭反应,气体产物经冷凝分水后,收集、检测。反应在设定时间结束时,关闭反应气体阀门,用N2吹扫,收集反应器内残存气体。待反应器内接触剂冷却后,取出并测定其上剩余焦炭含量。
当反应原料气为N2-O2混合气时,由于焦炭与O2的反应为强放热反应,为保持床层温度恒定,每次装入0.1g待生接触剂,并用9倍的同筛分惰性剂稀释;为排除反应时外扩散的影响,需加大气速以增加气体分子向待生接触剂颗粒表面的扩散速率。在相同温度下,固定氧分压(或水蒸气分压),测定不同床层气速下焦炭的转化率,结果表明,当床层气速大于0.025m/s时,转化率不再发生变化,这时可以认为消除了外扩散的影响。由于接触剂的粒径与常规FCC催化剂粒径相近,为很小的微球,因此认为颗粒内扩散阻力很小,边界层阻力可以忽略,化学反应速率为反应主要控制步骤。
1.3 产物分析方法
采用泰州市精密仪器有限公司TC-80C型红外定碳仪测定待生接触剂及气化反应后接触剂上的炭含量。采用北京分析仪器厂SP-3420型气相色谱分析仪(配备FID和TCD双检测器)归一化测定收集到的气体产物组成,用N2作载气。
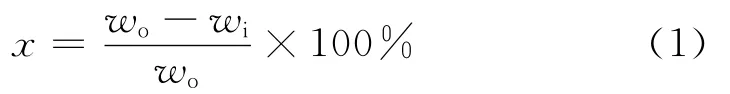
式(1)中,wo、wi分别表示待生接触剂和气化反应后接触剂上的焦炭质量分数,%。焦炭转化率x由式(1)计算。
2 结果与讨论
2.1 待生接触剂上焦炭与O2的气化反应规律
2.1.1 反应放热量与n(CO)/n(CO2)的关系
常规空气烧焦再生时,再生器内发生的反应较多,且具有不同的反应热,对过程热平衡产生不同的影响。S、N原子在焦炭中的含量相对很少,可忽略其反应热,笔者主要考虑C、H与O2的反应及其热效应,其主要反应方程式如式(2)~(6)所示。
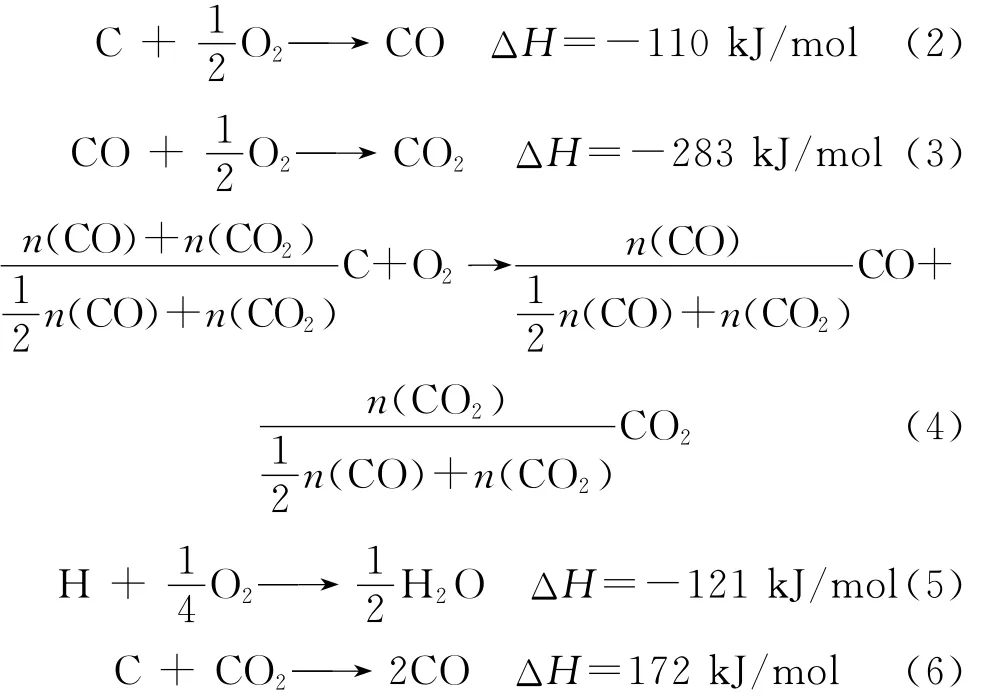
式(4)为C与O2反应的总反应式,n(CO)和n(CO2)分别为产物中CO和CO2物质的量。研究表明,式(6)所示在O2存在的情况下仍然会发生,但反应速率很小[13],为简化将式(6)所示反应忽略;焦炭组成为CHx,以x=0.8计,则1mol CH0.8与O2反应,C全部生成CO2时放热约为490kJ/mol,C全部生成CO时放热约为207kJ/mol。待生接触剂气化反应放出热量与产物气体中n(CO)/n(CO2)关系示于图2。从图2可见,气化反应放出热量随着产物n(CO)/n(CO2)的增加不断下降,且下降程度持续减小,并渐趋于平缓。未放出的热量储存于CO气体中,因此提高再生产物中n(CO)/n(CO2),可达到减少烧焦放热的目的。而CO作为费-托合成原料有较高的利用价值,同时也可减少再生时温室气体CO2的排放。
2.1.2 产物组成与反应温度的关系
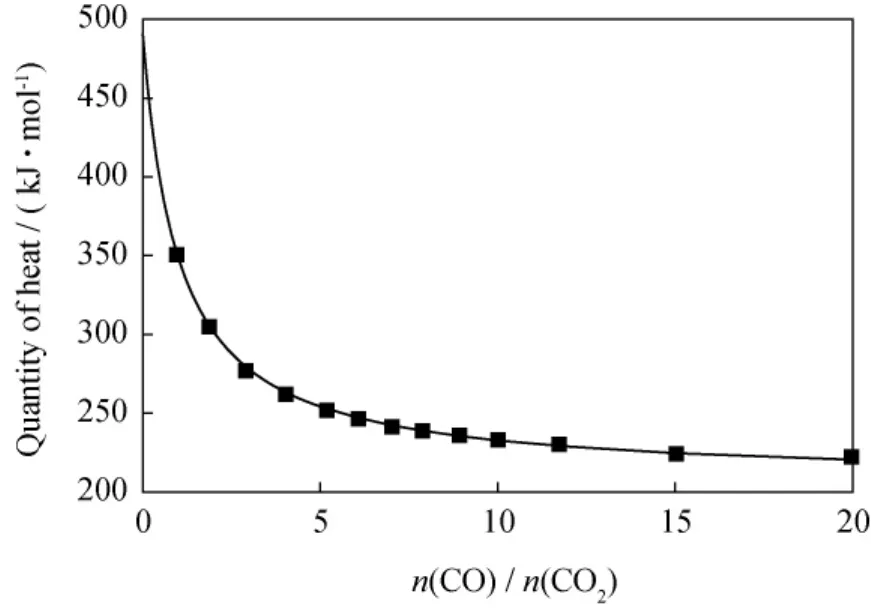
图2 待生接触剂上焦炭与O2反应放热量与气体产物中n(CO)/n(CO2)的关系Fig.2 Regeneration heat release vs n(CO)/n(CO2)in the products from the reaction of O2with coke on spent catalyst

图3 待生接触剂上焦炭与O2反应产物中CO的体积分数(φ(CO))和n(CO)/n(CO2)随反应温度(T)的变化Fig.3 CO volume fraction(φ(CO))and n(CO)/n(CO2)ofthe product vs Tfor the reaction of coke withO2on spent catalyst
图3给出了接触剂上焦炭与O2反应产物中CO体积分数和n(CO)/n(CO2)随反应温度的变化。由图3可见,在873~1073K范围内,随着反应温度的升高,n(CO)/n(CO2)先增加,973K后开始下降,此时产物中CO体积分数达到最大。这一结果与徐春明和燕青芝等[14-15]用FCC催化剂烧焦时所得结果相近。目前,关于焦炭与O2反应的研究较为普遍的结论是,焦炭与O2首先形成碳氧复合物COx,然后COx进一步分解生成CO与CO2,且二者同时发生[16-17]。相 关 研 究 表 明, 焦 炭 燃 烧 时 产 物 中n(CO)/n(CO2)有2个极值。在1000K以下,受化学反应控制,CO与O2发生均相氧化反应(见式(3))的机会较少,n(CO)/n(CO2)随着温度上升而增加;高于1000K时,CO与O2的均相反应加快,n(CO)/n(CO2)下降;随着温度的升高,反应逐渐受扩散控制,高于1250K时,反应被限制于接触剂颗粒外表面,生成的CO不再被进一步氧化,n(CO)/n(CO2)继续增加[18-20]。可以看到,在873~1073K间,笔者得到的实验结果与这些研究结果有较为相似的规律,但由于所使用的焦炭、实验装置有所不同,因此具体数据上存在一些差异。
将相位屏仿真的在轴闪烁指数结果与理论公式计算的在轴闪烁结果进行对比可得到图3所示结果,该理论结果与文献[15]相一致.图3为贝塞尔高斯涡旋光束在轴闪烁指数随光束宽度的变化情况,其中参数设置为从图3中可以得出贝塞尔高斯涡旋光束在轴闪烁指数相位屏仿真结果与理论计算结果相差无几,且随着光束宽度的增大,贝塞尔高斯涡旋光束的在轴闪烁指数变化呈现先增大后减小再增大的情况,其中w0≈0.012 m附近处的在轴闪烁指数呈现最大值,w0≈0.044 m附近处闪烁指数呈现最低值.
2.1.3n(CO)/n(CO2)与反应原料气中 O2体积分数的关系
图4分别给出了953和993K下,待生接触剂上焦炭与O2反应产物中n(CO)/n(CO2)与反应原料气中O2体积分数的关系。由图4看到,随着原料气中O2体积分数的增加,反应产物中n(CO)/n(CO2)不断下降,更多的CO被氧化生成CO2。因此,结合图3可以推断,使待生接触剂再生时减少放热,多生成CO的一个有效方法是将再生温度控制在973K左右;保证一定烧焦强度的同时,尽可能降低再生时原料气中O2的体积分数。
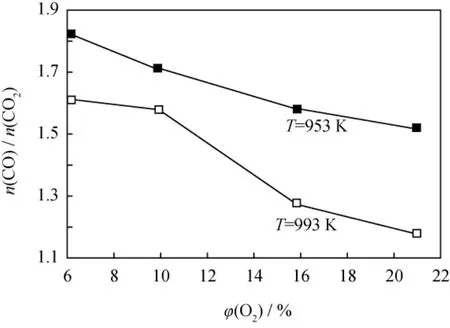
图4 不同反应温度下待生接触剂上焦炭与O2反应产物中n(CO)/n(CO2)随原料气O2 体积分数(φ(O2))的变化Fig.4 n(CO)/n(CO2)in the products vsφ(O2)of gasifying agent for the reaction of coke with O2on spentcatalyst at different temperatures
2.2 待生接触剂上焦炭与H2O(g)的气化反应的规律
在873~1173K范围、常压条件下,待生接触剂上焦炭与H2O(g)之间发生的主要反应如式(7)~(9)所示。
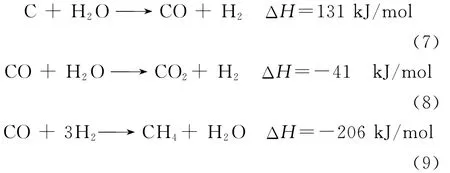
式(7)为水蒸气重整反应,它可以将焦炭转化成附加值较高的气体产物CO和H2。该反应为强吸热反应,极大地影响反应过程热平衡,1000K以下,该反应的化学反应平衡常数较低。式(8)为水蒸气变换反应,它影响气体产物中CO与H2之间的比例。式(9)为甲烷化反应,它是产物之间最重要的二次反应,通常易在较高压力和较低温度下发生。
2.2.1 产物组成和焦炭转化率与反应时间的关系
在反应温度1051K、气化剂中H2O(g)体积分数为98%条件下,待生接触剂上焦炭气化反应的产物组成及焦炭转化率随反应时间的变化示于图5。从图5可以看到,焦炭与H2O(g)反应的主要产物是CO、H2、CO2及少量的CH4。产物中CO+H2的体积分数随反应时间的增加平缓上升,CO2的体积分数不断减少,10min后,CO+H2体积分数达到90%后基本不变。反应前期CO的量大于H2的量,反应10min后H2的量大于CO的量,二者此消彼长的趋势是因为生成的CO在过量H2O(g)下进一步发生了水蒸气变换反应。从图5还可以看到,反应20min时,焦炭转化率达到60%左右。笔者推测,随着反应时间的继续增加,焦炭转化率会不断下降。这一方面是因为,待生接触剂上焦炭的实际组成是具有类石墨结构的稠环芳烃[21-22],随着反应的进行,稠环芳烃逐渐聚合、石墨化,焦炭反应活性不断减小[23],进而气化速率下降;另一方面,反应初期焦炭较为均匀地分布在接触剂的外表面,与H2O(g)能够较好地接触反应,而表面焦炭反应完之后,孔道内的焦炭与H2O(g)接触反应时,受内扩散影响而变得困难。
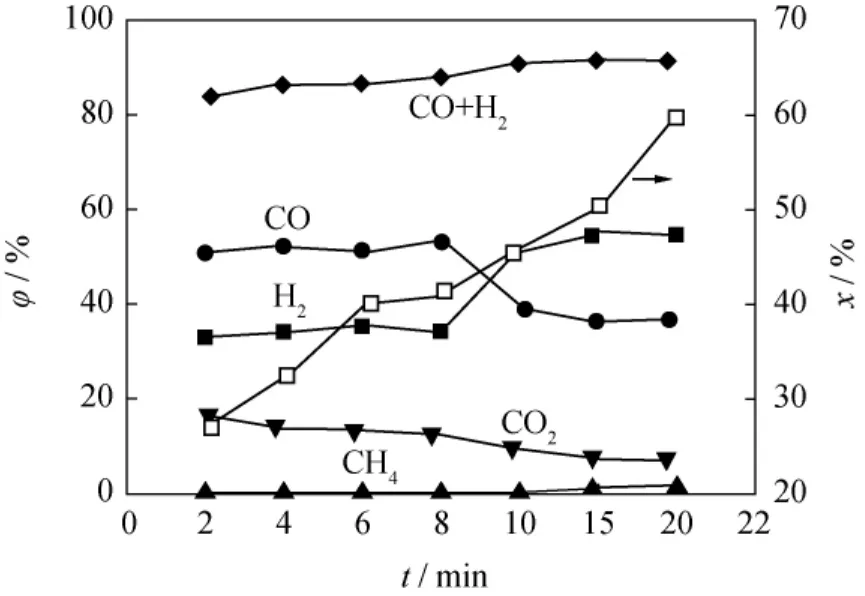
图5 待生接触剂上焦炭与H2O(g)气化反应产物体积分数(φ)及焦炭转化率(x)随反应时间(t)的变化Fig.5 Volume fraction of products(φ)and coke conversion(x)vs t in the reaction of coke with H2O(g)on spent catalyst
在反应温度1051K、反应时间10min条件下,待生接触剂上焦炭气化反应产物组成随气化剂中H2O(g)体积分数的变化示于图6。由图6可知,随着H2O(g)体积分数的增加,产物中H2体积分数先减小后增加,而CO体积分数正好相反,CO+H2体积分数随H2O(g)体积分数增加平稳上升,H2O(g)体积分数大于90%时,CO+H2的体积分数在80%以上;CO2体积分数随H2O(g)体积分数增加稳步下降,CH4体积分数基本处在0.6%~4.0%之间。

图6 待生接触剂上焦炭与H2O(g)气化反应产物体积分数(φ)随气化剂中 H2O(g)体积分数(φ(H2O))的变化Fig.6 Volume fraction(φ)of products vsφ(H2O)of gasifying agent in reaction of coke with H2O(g)on spent catalyst
2.2.3 产物组成及焦炭转化率与反应温度的关系
在100%H2O(g)为气化剂、反应时间25min条件下,反应温度对待生接触剂上焦炭气化反应产物组成及焦炭转化率的影响示于图7。从图7可以看出,随着反应温度的升高,产物中各气体体积分数变化较小,CO+H2体积分数维持在90%以上,CO2体积分数较低,处于4.5%~5.0%之间。反应温度对焦炭转化率的影响较为显著,反应温度高于1049K时,焦炭转化率明显增加,1122K时的焦炭转化率达到了90%以上。
2.3 待生接触剂上焦炭与O2-H2O(g)混合气气化反应的规律
2.3.1 产物组成及焦炭转化率与反应时间的关系
以O2-H2O混合气为气化剂进行的待生接触剂上焦炭的气化反应中,反应式(2)~(9)均存在,反应体系更加复杂,O2体积分数、反应温度对焦炭转化率及产物组成均有重要影响。

图7 待生接触剂上焦炭与H2O(g)气化反应产物体积分数(φ)及焦炭转化率(x)随反应温度(T)的变化Fig.7 Volume fraction(φ)of products and coke conversion(x)vs Tin the reaction of coke with H2O(g)on spent catalyst
在气化剂中O2体积分数为2%、反应温度1053K条件下,待生接触剂上焦炭与O2-H2O(g)混合气反应产物的组成和焦炭转化率随反应时间的变化示于图8。从图8可以看到,随着反应时间的增加,焦炭转化率迅速增加,7min时达到95%左右;产物组成中H2体积分数不断减小,6min后稳定在20%左右;CO体积分数平稳增加,5min后稳定在40%左右;CO+H2体积分数基本维持在60%左右;CO2体积分数保持在35%~40%间;CH4体积分数处于1%以下。

图8 接触剂上焦炭与O2-H2O(g)混合气气化反应产物体积分数(φ)及焦炭转化率(x)随反应时间(t)的变化Fig.8 Volume fraction(φ)of products and coke conversion(x)vs t in the reaction of coke with O2-H2O(g)on spent catalyst
2.3.2 产物组成及焦炭转化率与反应温度的关系
在O2-H2O(g)混合气中O2体积分数为1%、反应时间10min条件下,反应温度对待生接触剂上焦炭气化反应产物组成及焦炭转化率的影响示于图9。由图9可知,随着反应温度升高,产物中H2体积分数在20%~30%之间,且而先小幅下降后上升;CO体积分数变化恰与此相反,但幅度更小,基本维持在60%左右;CO+H2体积分数不断增加,1098K时达到84%左右;CO2体积分数不断减少,但基本没有生成CH4。这说明,气化剂中O2体积分数低有利于CO的生成,高温有利于H2的生成。1098K左右时,与采用H2O(g)气化剂相比,采用O2-H2O(g)混合气时,气化产物中CO+H2体积分数下降约5百分点,但CO增加量明显,H2的量较少。从图9还可以看到,反应温度高于1053K时,焦炭转化率在90%以上,进一步提高反应温度,焦炭转化率不再增加。由此推断,反应温度高于1053K时,焦炭与H2O(g)发生明显反应;低于此温度时,焦炭与H2O(g)的反应相对困难,主要发生的是焦炭与O2的反应。这一结果与涂永善[24]所得结果相似。

图9 待生接触剂上焦炭与O2-H2O(g)混合气气化反应产物体积分数(φ)及焦炭转化率(x)随反应温度(T)的变化Fig.9 Volume fraction(φ)of products and coke conversion(x)vs Tin the reaction of coke with O2-H2O(g)on spent catalyst
2.3.3 产物组成与气化剂中O2体积分数的关系
在反应时间3min、反应温度1033K条件下,待生接触剂与O2-H2O(g)混合气气化反应产物组成随O2体积分数的变化示于图10。从图10可以看到,随着混合气中O2体积分数的增加,焦炭气化产物中CO和H2的体积分数均呈不断减少的趋势,而CO2体积分数却不断增加;O2体积分数为1.0%时,产物中CO+H2体积分数达80%以上;当O2体积分数大于13.8%时,几乎没有H2生成。由于焦炭与O2的反应速率远大于焦炭与H2O(g)的反应速率[25],因此在O2充足的条件下,焦炭与O2的反应是主要反应,O2耗尽后发生焦炭与H2O(g)的反应;在O2过量的情况下,焦炭与O2-H2O(g)混合气气化反应产物组成与焦炭与O2反应的结果相当接近,产物中n(CO)/n(CO2)在0.25左右。

图10 待生接触剂上焦炭与O2-H2O(g)混合气气化反应产物体积分数(φ)随混合气中O2体积分数(φ(O2))的变化Fig.10 Volume fraction(φ)of products and coke conversion(x)vsφ(O2)in the reaction of coke with O2-H2O(g)on spent catalyst
3 结 论
(1)以O2对待生接触剂烧焦再生时,在873~973K范围内,产物中n(CO)/n(CO2)随温度升高不断增大;温度高于973K后,n(CO)/n(CO2)随温度升高不断下降,且随O2体积分数增加而下降。焦炭与O2反应生成CO时放出热量约为生成CO2时的1/3,因此通过调控反应温度和O2体积分数,可以控制气体产物中n(CO)/n(CO2),将过剩热量储存于CO中,一方面可达到调节系统热平衡的作用,另一方面可有效减少温室气体CO2的排放。
(2)采用H2O(g)对待生接触剂烧焦再生时,主要产物是CO、H2、CO2及少量的CH4。在反应温度1051K、气化剂中H2O(g)体积分数为98%条件下,产物中CO+H2体积分数达80%以上。反应温度对产物组成影响较小,但对焦炭转化率影响较为显著;反应温度高于1049K时,焦炭转化率明显提高;当反应温度为1122K、反应25min时,焦炭转化率达90%以上。
(3)采用 O2-H2O(g)混合气对待生接触剂烧焦再生时,反应温度越高,反应速率越大;在反应温度1053K、混合气O2体积分数为1.0%时,气体产物中CO+H2体积分数达到80%以上;O2体积分数高于10%时,产物组成类似于焦炭与O2反应所得产物组成。
[1]谷振生,王晓明.国内外重油加工技术新进展[J].炼油 与 化 工,2011,21(1):6-8. (GU Zhensheng,WANG Xiaoming.New progress of heavy oil processing technology at home and abroad[J].Refining and Chemical Industry,2011,21(1):6-8.)
[2]刘亚明.重油研究现状及展望[J].中国石油勘探,2010,15(5):69-76.(LIU Yaming.Current state and prospect of heavy oil research[J].China Petroleum Exploration,2010,15(5):69-76.)
[3]瞿国华.21世纪中国炼油工业的重要发展方向——重质(超重质)原油加工[J].中外能源,2007,12(3):54-62.(QU Guohua.Important orientation of China’s refining industry in 21th century—Processing heavy and super heavy crude oil[J].Sino-Global Energy,2007,12(3):54-62.)
[4]侯波,曹志涛.催化裂化工艺及催化剂的技术进展[J].化学工业与工程技术,2009,30(6):39-44.(HOU Bo,CAO Zhitao.Technology progress on process and catalyst of catalytic cracking[J].Journal of Chemical Industry &Engineering,2009,30(6):39-44.)
[5]王英杰,张忠洋,张玉.我国渣油加氢处理技术分析[J]. 当 代 化 工,2007,36(3):221-223.(WANG Yingjie,ZHANG Zhongyang,ZHANG Yu.Analysis on residue oil hydrotreating processing technology in China[J].Contemporary Chemical Industry,2007,36(3):221-223.)
[6]王雪松,袁志祥.延迟焦化工艺的技术进展[J].工业催化,2006,14(4):22-25.(WANG Xuesong,YUAN Zhixiang.Latest researches in the technologies for delayed coking[J].Industrial Catalysis,2006,14(4):22-25.)
[7]康建新,申海平.流态化焦化的发展概况[J].炭素技术,2006,25(3):28-33.(KANG Jianxin,SHEN Haiping.Development of fluid coking[J].Carbon Techniques,2006,25(3):28-33.)
[8]王洪亮,王刚,张东超,等.辽河减渣流化热转化预处理反应规律研究[J].现代化工,2010,30(增刊2):278-280.(WANG Hongliang,WANG Gang,ZHANG Dongchao,et al.Study on reaction rule of fluid thermal conversion pretreatment of Liaohe vacuum residue[J].Modern Chemical Industry,2010,30(Supply 2):278-280.)
[9]MAYES JR W W.Method of producing synthesis gas from a regeneration of spent cracking catalyst:US,6491810B1[P].2002-12-10.
[10]HETTINGER W P.Catalysis challenges in fluid catalytic cracking:A 49year personal account of past and more recent contributions and some possible new and future directions for even further improvement[J].Catalysis Today,1999,53(3):367-384.
[11]吴治国,张瑞驰,汪燮卿.炼油与气化结合工艺技术的探索[J].石油学报(石油加工),2005,21(4):1-6.(WU Zhiguo,ZHANG Ruichi,WANG Xieqing.The exploration of petroleum refining and gasification combined process[J].Acta Petrolei Sinica (Petrolem Processing Section),2005,21(4):1-6.)
[12]CORMA A,SAUVANAUD L,DOSKOCIL E,et al.Coke steam reforming in FCC regenerator:A new mastery over high coking feeds [J].Journal of Catalysis,2011,279(1):183-195.
[13]SANTOS L T D,SANTOS F M,SILVA R S,et al.Mechanistic insights of CO2-coke reaction during the regeneration step of the fluid cracking catalyst[J].Applied Catalysis A:General,2008,336(1-2):40-47.
[14]徐春明,林世雄,杨光华.裂化催化剂再生过程中密相床出口处CO2与CO比值的测定[J].石油大学学报(自然科学版),1990,14(6):84-92.(XU Chunming,LIN Shixiong,YANG Guanghua.Determination of CO2/CO in dense bed during cracking catalyst regeneration[J].Journal of China University of Petroleum(Edition of Nature Science),1990,14(6):84-92.)
[15]燕青芝,彭玉洁,程振民.催化裂化待生催化剂再生时产物中CO2/CO比的研究[J].石油炼制与化工,2001,32(4):56-58.(YAN Qingzhi,PENG Yujie,CHENG Zhenmin.Study on CO2/CO ratio in product gas of spent catalytic cracking catalyst during regeneration[J].Petroleum Processing and Petrochemicals,2001,32(4):56-58.)
[16]王光埙,林世雄,杨光华.裂化催化剂再生过程中的消碳动力学和机理[J].石油大学学报(自然科学版),1984,7 (3):268-274. (WANG Guangyun,LIN Shixiong,YANG Guanghua.Kinetics and mechanism of elimination of carbonaceous deposits on cracking catalysts in regeneration [J].Journal of China University of Petroleum (Edition of Nature Science),1984,7(3):268-274.)
[17]ZHU Z H,FINNERTY J,LU G Q,et al.A comparative study of carbon gasification with O2and CO2by Density functional theory calculations[J].Energy &Fuels,2002,16(6):1359-1368.
[18]LINJEWILE T M,GURURAJAN V S.The CO/CO2product ratio from the combustion of single petroleum coke spheres in an incipiently fluidized bed[J].Chemical Engineering Science,1995,50(12):1881-1888.
[19]ZENG T,FU W B.The ratio CO/CO2of oxidation on a burning carbon surface[J].Combustion and Flame,1996,107(3):197-210.
[20]SCALA F.A new technique for the measurement of the product CO/CO2ratio at the surface of char particles burning in a fluidized bed[J].Proceedings of the Combustion Institute,2009,32(2):2021-2027.
[21]张雪静,卢立军,徐广通.催化裂化待生剂上有机吸附物种的鉴别分析[J].分析化学,2006,34(增刊1):199-202. (ZHANG Xuejing, LU Lijun, XU Guangtong.Identification of the organic adsorptive species on fluid-bed catalytic cracking coking catalysts[J].Chinese Journal of Analytical Chemistry,2006,34(Supply 1):199-202.)
[22]杨雪,田辉平,王世环.催化裂化待生剂积炭结构组成的多重表征[J].石油学报(石油加工),2011,27(2):198-206.(YANG Xue,TIAN Huiping,WANG Shihuan.Characterization of coke on spent catalytic cracking catalysts by integrated techniques[J].Acta Petrolei Sinica (Petrolem Processing Section),2011,27(2):198-206.)
[23]刘鑫,张保申,周志杰,等.高温热处理对石油焦结构及气化活性的影响[J].石油学报(石油加工),2011,27(1):138-142.(LIU Xin,ZHANG Baoshen,ZHOU Zhijie,et al.Structure changes and gasification activity of petroleum coke after heat treatment[J].Acta Petrolei Sinica (Petrolem Processing Section),2011,27(1):138-142.)
[24]涂永善,罗雄麟,张秀英.水蒸气对裂化催化剂烧碳动力学的影响[J].石油大学学报(自然科学版),1995,19(5):71-74.(TU Yongshan,LUO Xionglin,ZHANG Xiuying.Effect of steam on the combustion kinetics of carbon in cabonaceous deposits on the cracking catalysts[J].Journal of China University of Petroleum(Edition of Nature Science),1995,19(5):71-74.)
[25]沙中兴,杨南星.煤气化与应用[M].上海:华东理工大学出版社,1995:32-33.
