RuO2/Al2O3催化甲醇选择氧化合成甲缩醛
2013-03-28陈文龙刘海超
陈文龙,刘海超
(北京大学化学与分子工程学院北京分子科学国家实验室,北京100871)
甲醇来源丰富,作为合成能源和化学品的重要平台分子,甲醇的转化利用得到人们的广泛关注[1,2]。作为甲醇的衍生物,甲缩醛(DMM)具有重要的工业应用价值,可以用于生产高浓度甲醛溶液[3]。同时,其毒性小、溶解性好、沸点低,也常用作制药和香水工业的溶剂。另外,甲缩醛具有高的氧含量和十六烷值,能100%溶解于柴油,因此,也可作柴油添加剂[4]。目前,工业上生产甲缩醛主要通过两步法,即甲醇首先在Ag或Fe-Mo催化剂上氧化脱氢转化为甲醛,然后甲醛与甲醇在酸催化剂作用下缩合生成甲缩醛。常用的酸催化剂有H2SO4,AlCl3以及酸性阳离子交换树脂等,对设备具有腐蚀性,而且两步法能耗高、工艺也较复杂,因此,近年来,甲醇一步氧化制备甲缩醛得到了人们的关注。甲醇一步氧化制备甲缩醛需要氧化中心和酸中心同时参与,即甲醇在催化剂氧化中心上氧化脱氢生成甲醛,然后甲醛与甲醇或催化剂表面甲氧基物种在酸中心上缩合生成甲缩醛[5,6]。Yuan等[7,8]发现含铼氧化物SbRe2O6和负载ReOx催化剂催化甲醇选择氧化产物中具有很高的甲缩醛选择性。随之人们相继报道Keggin杂多酸[5],Mo12V3W1.2Cu1.2Sb0.5Ox[9],硫酸化V-Ti-O[10]以及介孔Al-P-V-O[11]等双功能催化剂也具有较理想的甲缩醛收率。Liu等[6,12,13]发现负载RuO2催化剂具有优异的低温催化甲醇氧化反应性能,其产物选择性与载体表面的氧化还原性或酸碱性等密切相关,比如在ZrO2表面上,主要生成甲酸甲酯(MF),而在酸性Al2O3表面上,却有利于甲缩醛的生成[6]。本工作采用同时具有氧化中心和酸中心的RuO2/Al2O3催化剂,考察了不同Ru前体、制备方法和焙烧温度以及RuO2负载量等因素对催化剂结构及其催化性能的影响。
1 实验部分
1.1 催化剂的制备
载体γ-Al2O3由拟薄水铝石(德国Sasol)在500 ℃下空气中焙烧4 h获得。RuO2/Al2O3催化剂采用不同的方法制备。采用等体积浸渍法时,以RuCl3·nH2O(GR,国药化学试剂有限公司)、Ru(NO)(OOCH3)3(Alfa Aesar)或Ru(NO)(NO3)3(31.3% Ru,Alfa Aesar)为Ru前体,在常温下浸渍,然后经60 ℃干燥2 h,于120 ℃过夜干燥后,于500 ℃空气中焙烧4 h。采用均相氧化沉淀法(HOP method)时,按照文献方法[14],以RuCl3·nH2O为Ru前体,于80 ℃下用30% H2O2将Ru(Ⅲ) 氧化为RuO2,离心分离,并洗涤至用0.5 mol/L AgNO3在滤液中检测不到残余的Cl﹣,然后于120 ℃过夜烘干。采用沉积-沉淀法(DP method)时,以Ru(NO)(NO3)3为Ru前体,用氨水作沉淀剂调节溶液pH值为7~8,催化剂经过滤洗涤并于120 ℃过夜干燥,然后在所需焙烧温度下焙烧4 h。采用水热法制备RuO2/Al2O3催化剂时,按照文献方法[15],以RuCl3·nH2O为Ru前体,将RuCl3·nH2O和Al2O3置于水热釜中,150 ℃水热处理10 h,所得产物过滤,洗涤至用0.5 mol/L AgNO3在滤液中检测不到残余的Cl﹣,然后于120 ℃烘干。
1.2 催化剂表征
样品X射线粉末衍射(XRD)谱在Rigaku D/Max-2000型衍射仪上测定,管电压为40 kV,管电流为200 mA,采用Cu Kα(λ为0.154 06 nm)射线,扫描速率为3 (°)/min,扫描范围为20~60°或80°。催化剂粒径利用Scherrer公式计算得到,计算四方晶相RuO2粒径时采用(110)面对应衍射峰,计算γ-Al2O3粒径时采用γ-Al2O3(440)面对应衍射峰。透射电子显微镜(TEM)表征在Philips Tecnai F30 FEG-TEM上进行,加速电压为300 kV。测试前,将样品超声分散于乙醇中,静置后取少量上层溶液滴在具高分子薄膜的铜网上制样。扫描电镜(SEM)测试在Hitachi S-4800场发射扫描电子显微镜上进行,样品测试前进行喷金处理。催化剂表面组分的价态通过AXIS Ultra(Kratos, UK)能谱仪(XPS)表征,使用单色Al靶作为X射线源(Al Kα,hν为1 486.6 eV),测试功率为150 W(电压15 kV,电流10 mA),Ru3p电子结合能以载体Al2p结合能(74.4 eV)为标准进行校准。程序升温还原(H2-TPR)在TP5080多用吸附仪(天津先权)上进行,将含2.5 mg Ru量的催化剂放入石英反应管中,在流速为30 mL/min的5.0% H2/N2气流下以10 ℃/min从室温升至预定温度。H2消耗量使用热导检测器(TCD)检测,其中热导池温度为60 ℃,桥电流为120 mA。催化剂表面Ru的价态根据H2消耗量以商业CuO为标样标定计算。
1.3 催化剂评价
甲醇选择氧化反应在常压固定床微型反应器上进行。反应前,催化剂经造粒过筛为150~180 µm(80~100目),并用石英砂稀释以消除放热反应引起的局部过热现象。反应前催化剂预先在流速为30 mL/min的空气气氛下于300 ℃处理2 h。甲醇蒸气以鼓泡形式引入,反应原料气组成为3.0%CH3OH,20% O2,平衡气为N2。反应时,通过改变催化剂量和反应气流量以控制甲醇转化率约为20%,甲醇催化反应活性表示为每小时每摩尔Ru转化甲醇的物质的量(Activity,mol/(mol·h))。为避免产物冷凝,所有连接反应器和色谱的管线都保温在100 ℃以上。反应原料气和产物用Shimadzu GC 2010气相色谱在线分析,采用色谱柱为Porapak Q柱和5A分子筛填充柱,连接TCD检测器,反应稳定4 h后进行数据收集。在上述反应条件下Al2O3载体和石英砂上没有检测到产物的生成。
2 结果与讨论
2.1 Ru前体和制备方法对RuO2/Al2O3催化剂结构和催化性能的影响
表1列出了不同Ru前体和制备方法得到的质量分数为0.5% 的RuO2/Al2O3催化剂在120 ℃下的催化甲醇氧化反应活性及产物DMM、甲酸甲酯(MF)、甲醛(HCHO)和CO2的选择性。甲醇转化率影响其一级和二级反应路径[6],因此,在相近的甲醇转化率(约15 %)下,比较不同催化剂的甲醇氧化速率和产物选择性。如表1所示,RuO2/Al2O3催化剂制备过程中Ru前体的不同明显影响其催化反应活性。分别以RuCl3·nH2O,Ru(NO)(OOCH3)3或Ru(NO)(NO3)3为Ru前体,采用浸渍法制备的样品中,以Ru(NO)(NO3)3为前体制备的RuO2/Al2O3催化剂具有最高的催化反应活性,Ru(NO)(OOCH3)3次之,而以RuCl3·nH2O为前体制备的催化剂表现出很低的活性。
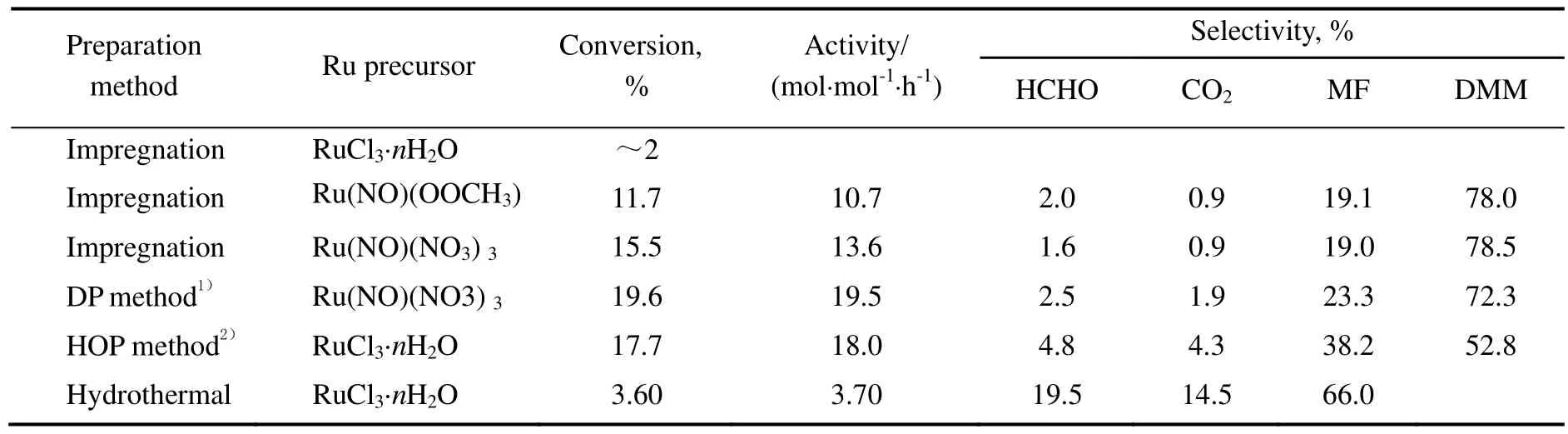
表1 不同Ru前体和制备方法对负载量为0.5%的RuO2/Al2O3催化性能的影响Table 1 Effects of Ru precursor or preparative method on RuO2/Al2O3 catalyst with 0.5% Ru loading for methanol selective oxidation
由表1可知,催化剂制备方法的不同也会影响其催化反应活性和选择性。采用沉积-沉淀法(DP)和均相氧化沉淀法(HOP)制备的RuO2/Al2O3催化剂较浸渍法和水热法得到的催化剂具有更高的催化反应活性。均相氧化沉淀法可以很好地分散RuO2,在载体表面RuO2以RuO2·xH2O的形式存在[14],故如图1中所示,在2θ 为 28.0,35.1和54.2°处没有观察到对应于四方RuO2的X射线衍射峰。
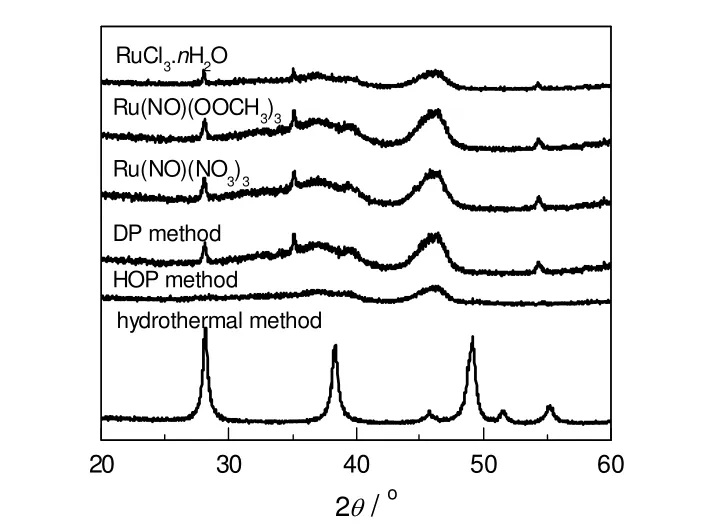
图1 不同Ru前体和不同制备方法制备的RuO2/Al2O3催化剂的XRD图谱Fig.1 XRD patterns for RuO2/Al2O3 catalysts prepared by different Ru precursors and different preparative methods

图2 不同焙烧温度下0.5% RuO2/Al2O3催化剂的XRD图谱Fig.2 XRD patterns for RuO2/Al2O3 catalysts calcined at different temperatures
然而,H2O2回流影响了Al2O3表面的酸性,导致均相氧化沉淀法得到的RuO2/Al2O3催化剂只有52.8%的甲缩醛选择性。Al2O3水热稳定性较差,因此,水热法制备催化剂过程中Al2O3转变为AlO(OH)(PDF: 49-01330),对应于XRD图中2θ为28.2,38.4和48.9°的衍射峰。所得到的RuO2/AlO(OH)催化剂催化甲醇选择氧化产物中以甲酸甲酯和甲醛为主,没有观测到甲缩醛的生成。综上所述,以Ru(NO)(NO3)3为Ru前体,采用沉积-沉淀法制备的RuO2/Al2O3催化剂具有较好的催化甲醇氧化反应活性和选择性。
2.2 焙烧温度对RuO2/Al2O3催化剂结构及催化性能影响
负载RuO2催化剂制备过程中焙烧温度对催化剂结构和催化反应性能具有重要的影响,如在ZrO2表面上,Huang等[13]发现焙烧温度的不同会导致载体表面高分散物种RuO42-构型的转变。图2为不同焙烧温度制得的RuO2/Al2O3催化剂的XRD图谱,表2中列出了由对应XRD结果计算得到的RuO2的粒径。由图2可知,随着焙烧温度从400 ℃升高到700 ℃,不同焙烧温度得到的质量分数为0.5%的RuO2/Al2O3催化剂结构保持不变,其中RuO2仍为四方晶相结构,载体Al2O3仍以γ-Al2O3形式存在。由表可知,当焙烧温度从400 ℃升到500 ℃时,RuO2粒径逐渐从21 nm增加到29 nm;在焙烧温度500~600 ℃下,RuO2的粒径基本保持不变;而当焙烧温度增加到700 ℃时,对应RuO2粒径则增加到35 nm。

表2 不同焙烧温度对0.5 % RuO2/Al2O3中RuO2粒径及催化甲醇选择氧化反应活性及选择性的影响Table 2 Effects of treatment temperature on average crystallite size of RuO2 in RuO2/Al2O3 and catalytic performance of RuO2/Al2O3 for selective oxidation of methanol to dimethoxymethane
表2还列出了在相近的甲醇转化率(约15%)下,焙烧温度对RuO2/Al2O3催化剂催化甲醇选择氧化活性和产物选择性的影响,其中产物选择性基本保持不变,甲缩醛具有约78%的选择性,甲酸甲酯选择性约为20%。随着催化剂的焙烧温度的增加,催化反应活性先增加后减少,当焙烧温度为500 ℃时,活性达到最高值。由XRD结果可知,焙烧温度大于500 ℃后,特别是直至650 ℃,RuO2平均粒径基本保持不变(约29 nm),这样催化反应活性的降低可能来源于更高温度下RuO2形貌的转变或RuO2与Al2O3载体之间相互作用的变化。
2.3 Ru负载量对RuO2/Al2O3催化剂结构及催化反应性能的影响
图3为不同Ru负载量的RuO2/Al2O3催化剂的XPS谱图,由于Ru3d结合能与C1s结合能位置重合,因此,本工作选用Ru3p来确定表面Ru物种的价态。由图3可知,在RuO2/Al2O3催化剂表面,尽管催化剂中Ru负载量不同,Ru3p3/2结合能均位于462.4 eV,Ru3p1/2结合能均位于484.8 eV,由此可以推断在不同负载量的RuO2/Al2O3催化剂表面Ru物种均以Ru4+的形式存在[17,18]。

图3 不同Ru负载量RuO2/Al2O3催化剂XPS图谱Fig.3 XPS spectra of RuO2/Al2O3 catalysts with different Ru loadings in the range of 0.30%-3.00% (mass fraction)
图4为不同Ru负载量的RuO2/Al2O3催化剂的XRD图谱。随着表面Ru负载量的增加,对应于RuO2的衍射峰强度不断增强。与负载氧化钒、氧化钼[19]以及RuOx/ZrO2[12]等催化剂不同,在Al2O3载体表面,当表面Ru负载量低至质量分数为0.30%时,仍可以观测到晶态RuO2的衍射峰,表明RuO2与载体Al2O3之间相互作用较弱。采用Scherrer公式计算得到的载体Al2O3以及RuO2的粒径见表3。从表可知,随着Ru负载量的增加,载体Al2O3的平均粒径基本保持不变,维持在4.5 nm左右,而RuO2平均粒径却逐渐从38 nm降到20 nm左右。
从XRD结果可知,不同负载量的RuO2/Al2O3催化剂中活性组分RuO2的平均粒径均远大于载体Al2O3的平均粒径。XRD反映的是催化剂体相结构的平均信息,采用TEM和SEM表征RuO2在Al2O3表面的存在状态,可以更好地认识RuO2在Al2O3表面的变化规律。图5为Ru负载量为1.00% 的RuO2/Al2O3催化剂的TEM图。从TEM图可看出,在Al2O3载体表面部分RuO2以纳米棒状结构形式存在,平均长度约为300 nm,平均宽度约为45 nm。HRTEM图中0.331 nm的晶面间距对应于RuO2(110)面,与XRD表征结果一致。
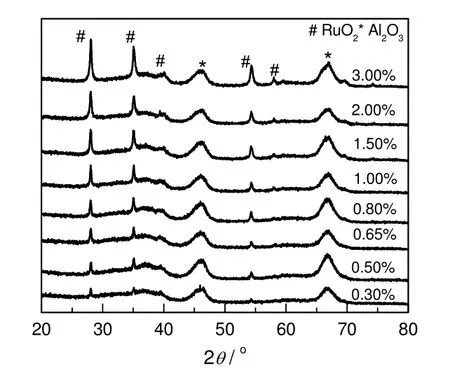
图4 不同Ru负载量RuO2/Al2O3催化剂的XRD图谱Fig.4 XRD patterns of RuO2/Al2O3 catalysts with different Ru loadings

图5 Ru负载量为1.00% 的RuO2/Al2O3催化剂的 TEM和HRTEM照片Fig.5 TEM and HRTEM images of RuO2/Al2O3 catalyst with 1.00% (mass fraction) Ru loading
图6给出了Ru负载量不同的RuO2/Al2O3催化剂的SEM图。由图可知:在载体Al2O3表面,当Ru负载量为0.30%时,RuO2主要以纳米棒状结构形式存在;当Ru负载量增加到1.00%时,开始出现不规则块状结构的RuO2,此时,纳米棒状结构与不规则块状结构RuO2同时存在,这与TEM结果相吻合;而当Ru负载量增加到2.00 %时,不规则块状RuO2成为RuO2的主要存在形式,其粒径大小不均匀,平均小于纳米棒状结构RuO2的粒径,这与XRD分析结果相一致。

图6 不同Ru负载量的RuO2/Al2O3催化剂的SEM照片Fig.6 SEM images of RuO2/Al2O3 catalysts with different Ru loadings (mass fraction)
图7为RuO2/Al2O3催化剂催化甲醇选择氧化活性和选择性随Ru负载量变化的规律。甲醛和深度氧化产物CO2在产物中所占比例均小于5%。随着Ru负载量增加,甲缩醛选择性从78.0%逐渐降低,当负载的Ru质量分数大于0.80%后稳定在72%左右,同时,甲酸甲酯选择性逐渐从17.9%升高至24%左右。随着Ru负载量的增加,催化甲醇氧化反应活性却逐渐从19.2 mol/(mol·h)降低到4.2 mol/(mol·h)。这些结果说明,尽管RuO2平均粒径逐渐减小,催化剂中Ru原子的本征催化反应活性逐渐降低,这与下面讨论的RuO2的氧化还原性降低相一致。

图7 不同Ru负载量RuO2/Al2O3催化剂催化甲醇选择氧化性能Fig.7 Effects of RuO2/Al2O3 with different Ru loadings on selective oxidation of methanol to dimethoxymethane Reaction conditions: conversion of CH3OH about 20%, 120 ℃,3.0% CH3OH, 20% O2, balance N2
甲醇选择氧化反应中,C-H键断裂生成甲醛是反应的决速步骤,反应遵循Mas-van Krevelen氧化还原机理,催化剂的催化反应活性与活性中心氧化还原性密切相关[6,20],因此,本工作采用H2-TPR考察了随着Ru负载量增加,RuO2/Al2O3催化剂中RuO2还原性的变化规律。不同Ru负载量RuO2/Al2O3催化剂的H2-TPR图谱如图8所示。

图8 不同Ru负载量RuO2/Al2O3催化剂的H2-TPR图谱Fig.8 H2-TPR profiles of RuO2/Al2O3 catalysts with different Ru loadings in the range of 0.30%-3.00% (mass fraction)
由图8可知,RuO2/Al2O3催化剂在H2-TPR图谱中主要存在两个还原峰,低温还原峰位于153 ℃附近,并且随负载量增加基本维持不变。而高温还原峰位于180 ℃附近,其随负载量的增加逐渐向高温位移,同时其在两个还原峰中所占的比例也逐渐增加。结合XRD,TEM和SEM等表征结果可知,低温还原峰可归属为纳米棒状结构RuO2的还原。随着负载量的增加,纳米棒状结构RuO2逐渐向不规则块状结构转变,因此,高温还原峰可归属为不规则块状结构RuO2的还原。由H2-TPR结果可知,纳米棒状RuO2较不规则块状RuO2具有更高的还原性。这与催化反应结果相吻合,即随着负载量增加所形成的不规则块状RuO2所占比例的逐渐增加,导致RuO2/Al2O3催化甲醇选择氧化反应活性逐渐降低。
由H2-TPR中H2的消耗量可以计算表面Ru物种的平均价态,计算结果列于表3中,表面Ru物种的平均价态为4.3左右,这与XPS分析结果Ru物种以Ru4+形式存在的结论相一致。由H2-TPR计算得到的平均价态略大于XPS分析结果,这可能与两种表征手段的误差或者氢溢流的发生有关。
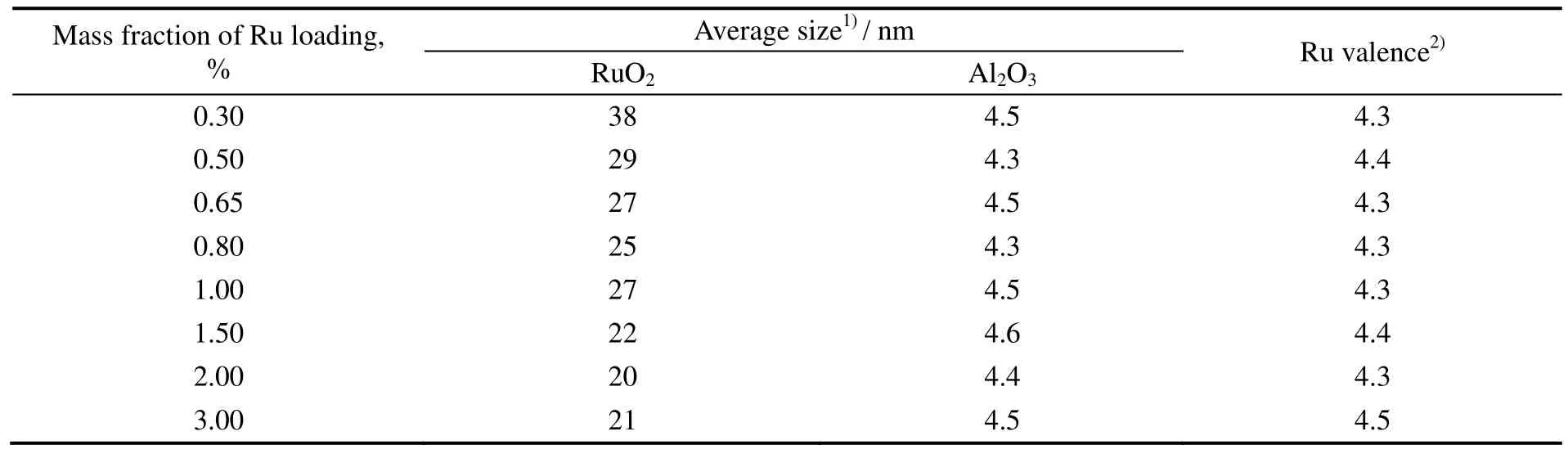
表3 RuO2/Al2O3催化剂中RuO2和Al2O3粒径以及依据H2-TPR计算的Ru的价态Table 3 Average crystallite size of RuO2 and Al2O3 or Ru valence on RuO2/Al2O3 catalysts with different Ru loadings
3 结 论
RuO2/Al2O3催化剂同时具有氧化中心和酸中心,在约20%的甲醇转化率下,其催化甲醇选择氧化产物中具有约80%的甲缩醛选择性。以Ru(NO)(NO3)3为Ru前体、通过沉积-沉淀法制备并经500 ℃焙烧得到的RuO2/Al2O3催化剂具有最优的催化反应性能。在Al2O3载体表面,随着表面Ru负载量的增加,表面RuO2逐渐由纳米棒状向不规则块状结构转变,其中低负载量时形成的纳米棒状RuO2具有较高的还原性和催化甲醇氧化反应活性。进一步提高RuO2/Al2O3催化剂催化甲醇选择氧化一步制备甲缩醛收率的关键是提高氧化中心RuO2的分散度以及载体表面的酸性等。这些认识为设计制备高效催化甲醇选择氧化合成甲缩醛的催化剂具有一定的指导意义。
[1] Olah G A, Goeppert A, Prakash G K S. Beyond oil and gas: the methanol economy[M]. Weinheim: Wiley-VCH, 2009: 1-350.
[2] Yang C J, Jackson R B. China’s growing methanol economy and its implications for energy and the environment[J]. Energy Policy, 2012,41: 878-884.
[3] Masamoto J, Iwaisako T, Chohno M, et al. Development of a new advanced process for manufacturing polyacetal resins Part 1:development of a new process for manufacturing highly concentrated aqueous formaldehyde solution by methylal oxidation[J]. Journal of applied polmer science, 1993, 50: 1299-1305.
[4] Ren Y, Huang Z H, Jiang D M, et al. Combustion characteristics of a compression-ignition engine fuelled with diesel-dimethoxy methane blends under various fuel injection advance angles[J]. Applied Thermal Engineering, 2006, 26(4): 327-337.
[5] Liu H C, Iglesia E. Selective one-step synthesis of dimethoxymethane via methanol or dimethyl ether oxidation on H3+nVnMo12-nPO40Keggin structures[J]. Journal of Physical Chemistry B, 2003, 107(39): 10840-10847.
[6] Liu H C, Iglesia E. Selective oxidation of methanol and ethanol on supported ruthenium oxide clusters at low temperature[J]. Journal of Physical Chemistry B, 2005, 109(6) : 2155-2165.
[7] Yuan Y Z, Liu H C, Imoto H, et al. Performance and characterization of a new crystalline SbRe2O6catalyst for selective oxidation of methanol to methylal[J]. Journal of Catalysis, 2000, 195(1): 51-61.
[8] Yuan Y Z, Shido T, Iwasawa Y. The new catalytic property of supported rhenium oxides for selective oxidation of methanol to methylate[J]. Chemical Communications, 2000, (15): 1421-1422.
[9] Royer S, Secordel X, Brandhorst M, et al. Amorphous oxide as a novel efficient catalyst for direct selective oxidation of methanol todimethoxymethane[J]. Chemical Communications, 2008, (7): 865-867.
[10] Fu Y C, Shen J Y. Selective oxidation of methanol to dimethoxymethane under mild conditions over V2O5/TiO2with enhanced surface acidity[J]. Chemical Communications, 2007, (21): 2172-2174.
[11] Chen S, Meng Y L, Zhao Y J, et al. Selective oxidation of methanol to dimethoxymethane over mesoporous Al-P-V-O catalysts[J].AICHE Journal, 2013, 59(7): 2587-2593.
[12] Li W Z, Liu H C, Iglesia E. Structures and properties of zirconia-supported ruthenium oxide catalysts for the selective oxidation of methanol to methyl formate[J]. Journal of Physical Chemistry B, 2006, 110(46): 23337-23342.
[13] Huang H, Li W Z, Liu H C. Effect of treatment temperature on structures and properties of zirconia-supported ruthenium oxide catalysts for selective oxidation of methanol to methyl formate[J]. Catalysis Today, 2012, 183(1): 58-64.
[14] Yu H, Zeng K, Fu X B, et al. RuO2·xH2O supported on carbon nanotubes as a highly active catalyst for methanol oxidation[J]. Journal of Physical Chemistry C, 2008, 112(31): 11875-11880.
[15] Xiang G L, Shi X J, Wu Y L, et al. Size effects in atomic-level epitaxial redistribution process of RuO2over TiO2[J]. Scientific Reports,2012, (2): 801, 1-6.
[16] Li W Z, Zhang H P, He X H, et al. Promoting effect of chloride ions on selective oxidation of methanol to methyl formate over zirconia-supported ruthenium oxide catalysts[J]. Journal of Energy Chemistry, 2013, 22(3): 512-516.
[17] Over H. Surface chemistry of ruthenium dioxide in heterogeneous catalysis and electrocatalysis: from fundamental to applied research[J].Chemical Reviews, 2012, 112(6): 3356-3426.
[18] Laursen A B, Gorbanev Y Y, Cavalca F, et al. Highly dispersed supported ruthenium oxide as an aerobic catalyst for acetic acid synthesis[J]. Applied Catalysis A: General, 2012, 433: 243-250.
[19] Gao X T, Wachs I E. Molecular engineering of supported vanadium oxide catalysts through support modification[J]. Topics in Catalysis,2002, 18(3-4): 243-250.
[20] Chen K D, Xie S B, Bell A T, et al. Structure and properties of oxidative dehydrogenation catalysts based on MoO3/Al2O3[J]. Journal of Catalysis, 2001, 198(2): 232-242.
