超声辅助酶切法用于溶液内蛋白质样品酶切特点分析
2013-03-15邹丽莉段进丹
成 洁,邹丽莉,段进丹,孙 伟
(中国医学科学院基础医学研究所北京协和医学院基础学院中心实验室,北京100005)
目前应用质谱进行蛋白质组研究需要将蛋白质酶切成多肽片段,再进行质谱分析,通过数据库检索实现蛋白质组的大规模鉴定[1]。但是在这个过程中,传统酶切法耗时长,往往需要16 h以上,极大的限制了蛋白质组分析的通量。超声辅助酶切法利用超声波辅助进行酶切,可以在几分钟内实现蛋白质的酶切,明显缩短了酶切的时间,增加了蛋白质组学研究的通量,是目前新开发的一种蛋白质快速酶切方法。虽然许多文献报道了应用超声辅助蛋白质组样品进行酶切[2-4],但超声辅助酶切法的优缺点,尤其是其所得到的多肽特性还有待进一步研究。本研究结合膜辅助溶液内酶切[5],通过与传统过夜酶切法比较,阐述超声辅助酶切法对溶液内蛋白质样品的酶切特点。
1 材料与方法
1.1 试剂
二硫苏糖醇(Dithiothreitol,DTT)、碘乙酰胺(Iodoacetamide,IAA)和血管紧张素Ⅱ(Sigma 公司);质谱级胰蛋白酶(Promega 公司);色谱级乙腈、甲酸、三氟乙酸和碳酸氢氨(Merk 公司);氯化钠,乙醇,盐酸等(北京化学试剂公司)。牛血清白蛋白(Bovine serum albumin,BSA)(北京鼎国生物技术有限公司)。
1.2 蛋白质酶切
本实验应用标准蛋白BSA 进行两种方法的比较,两种酶切方法分别重复3 次,结果用均数±标准差(±s)表示。BSA 以1 g/L溶于裂解液(7 mol/L尿素,2 mol/L 硫脲,65 mmol/L DTE,100 mmol/L Tris)中。两支10 ku套管分别取200 μg BSA,加入DTT 使其终浓度为20 mmol/L,37 ℃水浴1 h,离心除去液体并用UA(8 mol/L 尿素,0.1 mol/L Tris/HCl pH 8.5)清洗样品;加入50 mmol/L IAA 溶液,室温避光45 min,离心除去液体并用UA 冲洗后,加入200 μL 25 mmol/L碳酸氢铵,以1∶20的比例加入胰蛋白酶,分别用传统过夜酶切法和超声辅助酶切法进行酶切[5]。传统过夜酶切法将样品置于37 ℃水浴13 h后,再以1∶50比例加入胰蛋白酶,继续水浴3 h后完成酶切。超声辅助酶切法用2 mm超声破碎仪探头伸入溶液中进行酶切,超声功率20 W,工作时间60 s,暂停30 s,两个循环后,再以1∶50比例加入胰蛋白酶,超声两个循环,完成酶切(表1)。得到的多肽用C18 柱萃取后真空抽干。
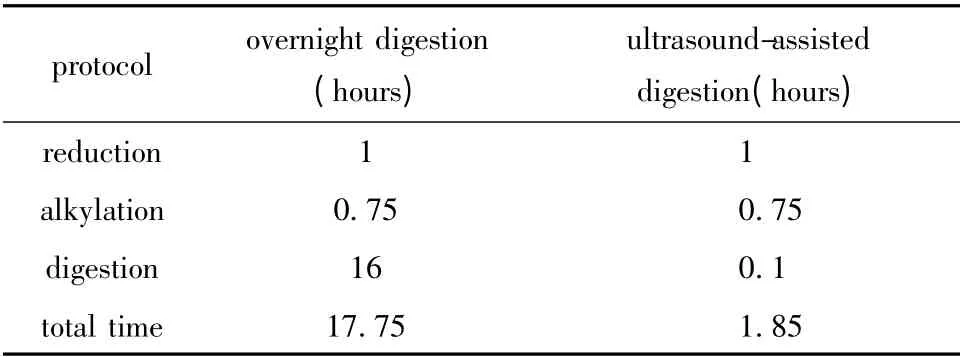
表1 传统过夜酶切法和超声辅助酶切法的比较Table 1 The comparison of overnight and ultrasoundassisted digestion methods
1.3 多肽浓度的检测
酶切后的多肽样品重溶于1‰甲酸后应用BCA法测定多肽浓度。血管紧张素II 配成1、0.5、0.25、0.1、0.05 及0 g/L的标准品,将10 μL不同的浓度的标准品及多肽样品分别加入96 孔板中,再加入200 μL配好的工作液,显色后测其在波长562 的吸光度。根据标准品的浓度及其相应的吸光度值制作浓度/吸光度标准曲线,计算出样品浓度。
1.4 一维胶分离
酶切前后BSA 样品加入还原剂和上样缓冲液,在100 ℃条件下加热5 min,应用4% ~12% SDS 胶分离,恒压200 V,时间为35 min。应用微波辅助方法进行考马斯亮蓝染色[6],Alpha 凝胶成像系统成像。
1.5 质谱分析
将1 μL酶解液点入384 孔MALDI-TOF 靶中,待样品自然干燥后,加入a-Cyano-4-hydroxycin-namie acid 饱和溶解基质(70% ACN,0.1% TFA),同时在校准点内加入已知质量数的多肽混合物作为外标校准物。质谱方法采用反射正离子模式,20 KV加速电压,激光强度为1%,扫描质量范围0.5 ~3.5 ku。所得质谱图用MASCOT(V2.3.02)软件检索,所用数据库为牛的蛋白质数据库(ftp.ncbi.nih.gov/)。检索参数为:胰蛋白酶,误切位点为2,母离子质量误差为50 ppm,固定修饰为脲甲基化修饰Carbamidomethy(C),可变修饰为氨基甲酰化修饰Carbamy(N-term)。
2 结果
2.1 传统过夜酶切法与超声辅助酶切法的多肽回收率
传统过夜法的多肽回收率为47% ±3.9%,超声辅助酶切法的多肽回收率为39% ±2.7%,较传统酶切法低8%。
2.2 传统过夜酶切法与超声辅助酶切法酶切后一维胶结果
酶切前后的BSA 样品应用SDS 胶进行分离,结果显示两种方法酶切后的条带相似,在BSA 位置的蛋白条带消失,在3.5 ku附近出现了酶切后的多肽条带(图1)。
2.3 传统过夜酶切法与超声辅助酶切法酶切后飞行时间质谱结果
两种方法酶切得到的肽段进行了基质辅助激光飞行时间质谱仪分析。图2A 与图2B 图分别为两种方法得到的BSA 肽指纹图谱。用MASCOT 软件对质谱图进行了数据库检索,结果显示(表2),超声辅助酶切法相比于传统过夜酶切法的MASCOT 评分高122.8%,匹配的多肽数多47.7%,肽段的覆盖率高10%,误切率高22%。
2.4 传统过夜法与超声辅助酶切法酶切后多肽特征的分析
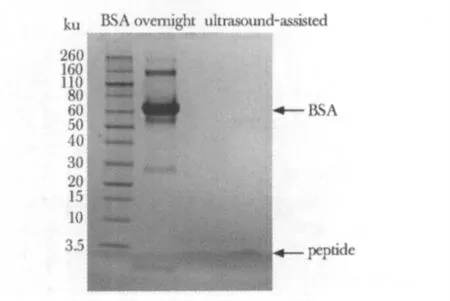
图1 传统过夜法与超声辅助酶切法酶切BSA前后一维胶结果Fig 1 One-dimensional gel of BSA before and after digestion using overnight and ultrasoundassisted methods
将在3 次重复实验中均得到鉴定的多肽进行分析(表3),传统过夜酶切法3 次重复鉴定的多肽有36 个,超声辅助酶切法有49 个,两种方法共有的多肽为28 个(图3A),超声辅助酶切法得到的多肽比过夜酶切法多13 个,并包含了过夜酶切法匹配肽中78%的肽段。
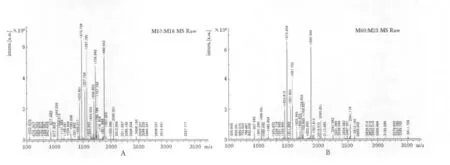
图2 两种酶切方法飞行时间质谱结果Fig 2 The MALDI-TOF mass spectra of BSA from overnight and ultrasound-assisted methods
表2 两种方法所得到的质谱鉴定结果Table 2 The MS results from overnight and ultrasound-assisted methods(±s,n=3)

表2 两种方法所得到的质谱鉴定结果Table 2 The MS results from overnight and ultrasound-assisted methods(±s,n=3)
*P<0.01 compared with control.
methodmascot scoreNo.of peptidesSequence coverage (%) miscleavage peptide rate (%)overnight digestion101 ±28*42 ±5*71% ±6%38% ±9%*ultrasound-assisted digestion225 ±16*62 ±5*81% ±4%60% ±5%*
对两种方法共57个多肽的误切位点的分析表明(图3B),共有的28 个肽段误切率为29%,只被过夜酶切法鉴定的多肽误切率为37%,只被超声辅助酶切法鉴定的多肽误切率为95%,明显高于过夜酶切法。
对上述57 个多肽的分子量(图3C)及多肽长度(图3D)分布情况分析表明,在分子质量小于2 ku、长度小于20 个氨基酸时,两种方法得到的肽段数目无明显差别。但在分子质量大于2 ku和长度大于20 个氨基酸时,超声法获得的肽段数量明显高于传统方法,而且这些肽段大多有胰蛋白酶的误切位点。
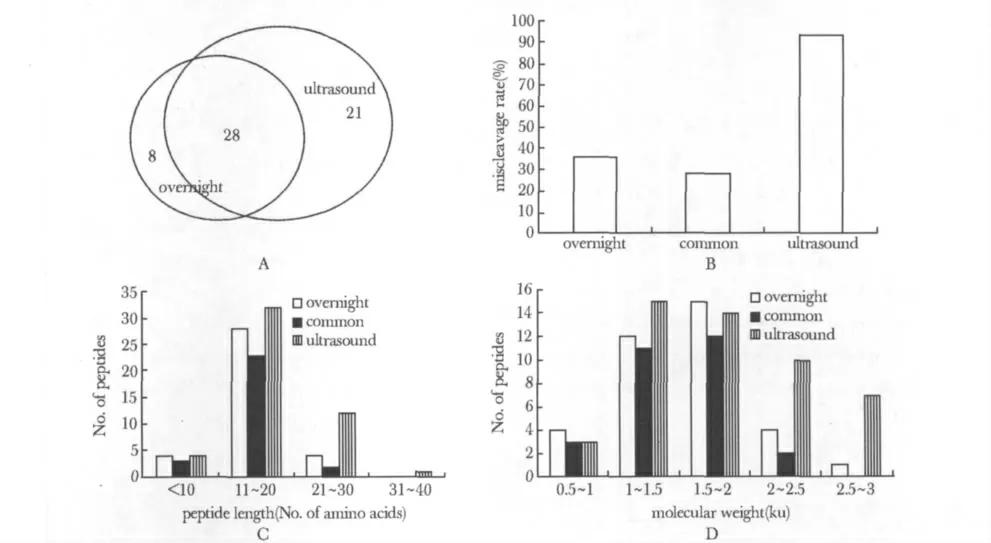
图3 两种方法得到多肽性质分析Fig 3 The analysis of peptides from two methods
3 讨论
超声辅助酶切法是利用超声波进行快速酶切的一种方法,其可以明显加快蛋白质的酶解过程,提高蛋白质组学的分析速度。虽然其具体工作原理还不清楚,但其酶切效果得到了众多研究成果的证实。目前这方面的应用研究很多,但超声辅助酶切法对溶液内蛋白质样品的酶切优缺点,尤其是鉴定出多肽的特点还没有报导,因此本文通过与过夜酶切法方法的比较,阐述了其酶切的特点。
本研究通过对标准蛋白BSA 溶液内样品的酶切,证明超声辅助酶切法可以在10 min之内完成酶切。通过质谱分析结果表明,其酶切出的多肽数要高于过夜酶切法,因此得到了更高的MASCOT 得分和蛋白覆盖率,说明超声辅助酶切法可以得到更好的质谱鉴定结果,上述结论与以往报导相一致。
进一步对其鉴定出的多肽性质分析表明,超声辅助酶切法得到的多肽误切率明显高于传统过夜酶切法,而且比过夜酶切法多鉴定出的多肽大多含有误切位点,说明该方法酶切不完全,其可以得到更多的多肽和更高的蛋白质覆盖率是由不完全酶切造成的,这也可以部分解释其酶切后多肽的回收率要低于传统过夜酶切法。
在蛋白质组学应用中,应用超声辅助酶切法可以得到更多的肽段和更高的蛋白质覆盖率,因此可以用于发现蛋白质组学的分析,可以鉴定到更多的蛋白或多肽,尤其是一些低丰度蛋白或用传统过夜方法难以酶切的蛋白。另一方面,超声辅助酶切法对蛋白质的酶切不完全,会增加样品的复杂程度,增加靶向分析的多肽检测难度,因此不太适用于靶向蛋白质组学分析。
[1]Yates JR.Mass spectral an alysis in proteomics[J].Annu Rev Biophys Biomol Struct,2004,33:297-316.
[2]Filip D,Pavel B,Karel M,et al.Rapid and efficient protein enzymatic digestion:An experimental comparison[J].Electrophoresis,2012,33:288-295.
[3]Carrera M,Canňas B,López-Ferrer D,et al.Fast monitoring of species-specific peptide biomarkers using high-intensity-focused-ultrasound-assisted tryptic digestion and selected MS/MS ion monitoring[J].Anal.Chem,2011,83:5688-5695.
[4]Kirk CH,Lauren K,Ori M,et al.An in-solution ultrasonication-assisted digestion method for improved extracellular matrix proteome coverage[J].Mol Cell Proteomics,2009,8:1648-1657.
[5]Jacek RW,Alexandre Z,Nagarjuna N,et al.Universal sample preparation method for proteome analysis[J].Nat Methods,2009,6:359-362.
[6]Nesatyy VJ,Dacanay A,Kelly JF,et al.Micorwave-assisted protein staining:mass spectrometry compatible methods for rapid protein visualization[J].Rapid Commun Mass Spectrom,2002,16:270-280.
