超分辨光学显微镜的生物学应用
2013-02-24毛秀海杜建聪樊春海邓素辉
毛秀海 杜建聪 黄 庆 樊春海 邓素辉
(中国科学院上海应用物理研究所嘉定园区 上海 201800)
超分辨光学显微镜的生物学应用
毛秀海 杜建聪 黄 庆 樊春海 邓素辉
(中国科学院上海应用物理研究所嘉定园区 上海 201800)
生物学家一直渴望使用无损、可实时成像的远场光学显微镜对细胞内动力学行为或者纳米尺度的蛋白质-蛋白质相互作用进行研究。由于光学衍射极限的存在,传统的远场光学显微镜无法对200纳米尺度内的这些生命活动进行观察。近年来,克服光学衍射极限的超分辨成像技术快速发展,将分辨率提高至数十纳米级别。基于超分辨技术的荧光显微镜包括受激发射损耗显微镜(STED)、光敏定位显微镜(PALM)以及随机光重建显微镜(STORM)。这些技术日趋成熟并成功应用于细胞生物学、微生物学、神经生物学等研究,为整个生命科学的发展带来全新的认识。
光学显微镜,超分辨,生物学,纳米,蛋白质
生物学研究往往需要在活细胞条件下观察胞内组织的活动。光学技术因其具备对生物样品扰动最小、实时可见的优势,从而成为生物学观察的重要技术手段[1–4]。远场光学显微镜一直促进着人类对微观世界的理解,并伴随着整个生物学领域的产生和发展。三百多年以前,Hook等[4]对成像原理进行深入研究,发明了最早的光学显微镜。利用他发明并制作的光学显微镜首次观察到细菌以及微生物,开创了生物学领域的新研究。常规的光学显微镜的横向分辨率约200 nm,纵向分辨率400–700 nm,因此,无法观测纳米大小的许多重要的生命体,如病毒和细胞,以及生物体内的蛋白质等都无法进行观测。为了揭示细胞内蛋白质活动和细胞微结构特征,提高光学显微镜分辨率成为细胞生物学发展的迫切要求。
1 超分辨成像的重要技术进展
近20年来,克服光学衍射极限的超分辨显微镜不断发展,基于荧光的显微镜技术利用分子的荧光能级跃迁特性、荧光灵敏度高和特异性好的特点,将显微镜分辨率提高至纳米尺度。超分辨技术的基本原则是减少或避免处在激发体积内的分子同时发射荧光。目前,超分辨显微成像可以分为两种方法。第一类是结合光学非线性效应对成像的照明光路进行整形,获得小于衍射极限的荧光发光点。这类技术的典型代表是Hell等[5]提出的受激发射损耗显微镜(Stimulated emission of depletion microscopy, STED)。同时,STED也是第一个用来突破衍射极限的远场光学显微技术,其原理是采用两束组合激光,一束激光被聚焦成正常的衍射极限焦斑,使焦斑内的荧光分子处于激发态;另一束为中心光强为零环形焦斑分布的损耗光,两束光进行叠加,损耗光通过受激发射过程损耗周边区域内的激发态荧光分子。因此,周边区域内的荧光分子被淬灭,只剩下中心的荧光发光点,从而实现小于衍射极限的荧光发射面积。从原理上讲,由于损耗光的中心光强为零,只要淬灭光足够强,则由第一束光激发的荧光分子所占的体积可以被压缩到极小的范围内,极大提高荧光显微镜的分辨率。目前,STED显微镜可实现20 nm分辨率的免疫荧光成像和50–70 nm的荧光蛋白成像[6–17]。
第二类超分辨技术是基于单分子定位的成像方法,利用光开关荧光蛋白,光敏化或光漂白现象将衍射极限范围内的单个分子在不同的时间随机地激活,并将各个荧光分子精确定位再重组,叠加获得超分辨图像。该类技术通过将相互位置过于靠近而无法被传统光学显微镜同时分辨的荧光标记分子逐个予以分别激发,使各个荧光分子所成的艾里斑像之间不再相互干扰,从而能够对每个独立的荧光分子逐个进行定位。这样就可以通过提高时间分辨率,来达到改善空间分辨率的目的。这一类技术的典型代表有光敏定位显微镜(Photoactivation localization microscopy, PALM)和随机光重建显微镜(Stochastic optical reconstruction microscopy, STORM)等。
2006年,Betzig等[18]首次使用光敏绿色荧光蛋白作为探针标记样品,利用该光敏绿色荧光蛋白只有在405 nm的激光敏化后,才能被561 nm激发发射绿色荧光。他们首先利用低能量的405 nm激光辐照,仅敏化极稀少的光敏蛋白,再使用561 nm激光进行激发直至被记录的分子发生光漂白。重复敏化-激发-定位-漂白过程,完成足够多的分子被记录。这种技术被称为光敏定位显微镜(PALM)。文献[19–29]中均介绍了该技术成果是2006年重要的研究发现,入选了2006年《Science》十大进展。
STORM成像方法与PALM类似,哈佛大学的Bates等[30,31]提出利用光控荧光分子可实现超分辨成像,即通过激光控制分子在荧光态和非荧光态之间转换。实验中,他们将Cy3和Cy5分子一同修饰在抗体上来标记细胞内的蛋白,用532 nm波长的激光敏化探针,632 nm波长的激光来观察、定位,重复绿光开启-红光激发-定位-绿光开启过程,记录所有分子的位置,叠加组成一幅完整的图像。在活细胞成像中,使用明暗转换快速且较亮的探针标记样品,可以让2D成像的空间分辨率达到25 nm,而时间分辨率最快可达到0.5 s。与PALM相比,虽然两种成像方法精度相同,但STORM可以重复记录,对荧光分子随机运动的影响更小[32–39]。
2 超分辨技术应用
近几个世纪以来,远场光学显微镜一直促进着人类对微观世界的理解,并伴随着整个生物学领域的产生和发展。尽管许多超分辨方法已经在原理上实现了小于衍射极限的成像效果,但其中很大一部分技术距离生物学应用较远,需要进一步探索和发展。在这些超分辨技术中,STED、STORM和PALM发展较早也日趋成熟,目前已经广泛应用于生物领域的研究,给生物学发展带来了一些全新的发现。
2.1超分辨技术在细胞生物学中的应用
随着超分辨荧光显微镜的发展,很多亚细胞结构,例如微管、肌动蛋白和线粒体等,被选为标准模型来验证超分辨技术的分辨率和可靠性。这些研究不仅展现超分辨荧光显微镜的高分辨率,也说明该技术可以被用于研究分子尺度的细胞结构及相关功能。其中,胞膜蛋白以及胞膜微区的研究是超分辨技术应用的重点和难点。因为这些结构太小,无法用常规的显微镜观察。而超分辨荧光显微镜的超分辨能力可以解决这些问题。例如,在细胞膜研究中,之前用生物化学的方法测得线粒体膜上的阴离子通道(hVDAC)是与细胞质基质中的己糖激酶-1相结合的。但Neumann等[14]用双色STED荧光显微方法观察线粒体的结构,他们发现大部分的hVDAC并没有与在线粒体上的己糖激酶-1相结合。这些研究充分展示了超分辨技术可以突破生物领域的瓶颈,有助于进一步认识细胞各蛋白的结构与功能。
PALM荧光显微技术也提供新的视角来观察质膜中的蛋白组织以及胞内细胞器。Bakshi等[40]使用该技术得到RNA聚合酶在大肠杆菌中的分布及扩散过程。同时English等[41]通过实时定位ReIA 分子的扩散活动,发现蛋白的活性随着分裂周期而发生变化。Matsuda等[24]在染色质的研究中,观察到染色质纤维结构。为了进一步提高PALM数据的空间分析能力,Burnette等[36]联合应用PALM荧光显微技术和双色荧光分子标记方法,以提高成像的清晰度。并且该新方法被广泛用于细胞受体之间相互作用的研究中。例如Betzig等[18]在研究细胞粘附蛋白的动力学过程中,用该技术准确定位两种不同蛋白在膜上的相对位置。例外,Sherman等[42]研究了T-淋巴细胞与TCR(T-Cell antigen Receptor)结合过程(图1)。他们清晰地观测到在T-淋巴细胞膜上的一系列蛋白中间体和聚集体的结构变化,并且发现TCR信号分子存在纳米尺度的功能区。这一结果扩展人们对T细胞活化机制以及TCR蛋白介导的信号复合物的形成和组织的理解。因此,PALM荧光显微技术不仅可以突破常规方法的极限、清晰观测分子尺度的细胞结构,而且为研究蛋白间的动态相互作用提供了更加便捷和可靠的途径。

图1 T-淋巴细胞与TCR结合的PALM成像Fig.1 PALM imaging of interaction between T-Cell and TCR.
STORM荧光显微镜也被用于观察分子尺度的亚细胞结构。其中Bates等[31]利用STROM观察了细胞BS-C-1的线粒体网络结构,解析了观察微管和网格蛋白包被单元。并且证明STORM具有相当高的分辨能力,可以突破普通荧光显微镜的极限,将分辨出重叠或者不能分解的细丝。而Huang等[34]进一步改进了超分辨技术,并发明新型的3D STORM显微技术。他们结合使用共聚焦平面三维扫描技术和折射率错配修正方法获得了20–30 nm的横向分辨率和50–60 nm的纵向分辨率。并且得到了清晰的细胞微管成像,也证明线粒体具有纤薄外壳以及内部空心的结构(图2)。他们提出根据线粒体外壳形态,可将与线粒体连接的微管分为细胞内球状分散结构和管状交联结构两类。同时他们观察到线粒体与微管之间存在尺蠖式的相互接触模式。这为线粒体的研究及其运输机理提供了新的视角,从而进一步研究细胞内的活动与功能。
这些结果展示了超分辨荧光显微镜的发展,推进了细胞生物学的研究。生物学家们期待着用超分辨显微镜观察到更多的细胞亚结构。

图2 细胞微管的STORM成像Fig.2 3D STORM image of the mitochondrial network in cell.
2.2超分辨技术在神经生物学中的应用
自从在一个世纪之前用光学显微镜观察到神经细胞后,人们已经知道大脑主要的工作方式是通过将信息从神经元中的轴突传送到树突。而神经元的性质与突触结构息息相关。为研究神经细胞的性质和功能,需要了解突触的分子尺度的结构及其性质。实际上,突触的功能是由一个含有数百种蛋白的蛋白工厂精确调控的。研究突触的功能和结构需要对树突位点上的蛋白进行精确的定位和研究。因此,完成神经细胞的研究需要既能满足纳米尺度的分辨率,又可以与活体成像相容的方法。而超分辨荧光显微镜可以满足这些苛刻的要求。
Meyer等[43]用STED荧光显微技术发现AMPA受体在神经细胞的突触上形成一个环形的分布。Nagerl[11]则用STED显微技术展现了细胞中的突触顶端的形状和结构。Ding等[13]用双光子STED显微技术观察在深层组织样品中的突触顶端的形态。其中Van等[44]通过超分辨的STED荧光显微技术,清晰地观测到在胞吐作用中起重要作用的受体蛋白syntaxin会因静电相互作用而被阴离子脂类PIP2紧紧包裹,并形成72 nm大小微区(图3)。同时,他们也发现在缺少胆固醇时,PIP2与受体蛋白syntaxin仍能形成这种微区。这个结果说明磷脂双分子层中的微区主要是通过蛋白与脂类的静电作用而形成。这些结果指出蛋白在细胞膜中的活动与在胞质的活动的机理是不同的。

图3 细胞膜成像技术Fig.3 Confocal and corresponding STED image of membrane.
除了研究在神经元内的亚细胞结构,超分辨显微镜也被用来研究神经细胞的形态。例如,Willig等[7]在突触结合蛋白的研究中,发现这些蛋白在胞吐作用后仍处以团簇状态。Hell等[45]成功地利用STED显微镜观测活鼠大脑皮层神经元及其精细的动力学过程,实现了高速检测活细胞内高分辨率图像,并且分辨率已经提高至数十纳米,远远突破了光学衍射极限。STED荧光显微技术为以后神经间相互作用的研究,提供了可靠的方法。
STORM也被用于研究突触的分子尺度的构型。例如,Dani等[46]在神经轴突的研究中,利用该技术成功定论地研究了神经递质在神经轴突传递的方式以及神经突触在神经细胞上的分布,并且对突触中神经递质的组成成分进行了定量分析。同时随着STORM荧光显微技术不断发展,3D多色STORM的方法取得了诸多成功,并成功用于研究神经细胞中微丝和血影蛋白的组织结构中。Xu等[47]使用多色STORM荧光显微技术观测到微丝蛋白在轴突中形成环状结构(图4),同时发现每两个微丝蛋白环之间由一个血影蛋白连接,并构成一个周期性的结构。他们发现该环状结构与轴突中的钠离子通道有一定关联,而在树突中,没有这种周期性的结构,而是有较长的微丝蛋白存在。

图4 神经突触的3D STORM成像Fig.4 3D STORM image of actin in a dendritic region.
达到数十纳米的分辨率可以研究分子级别的组织和细胞器,而几个纳米的分辨率就可以达到直接观察分子与分子间反应的要求。尽管可以用不同的超分辨显微镜得到数十纳米的分辨率,但由于在追踪轴突在脑神经的分布却需要几个纳米的分辨率,因此仍需要更高的分辨率。随着现有超分辨技术的发展,在亚神经元的研究中会得到更直观的结果。
2.3超分辨技术在微生物学中的应用
近几年的研究中,对于细菌结构的认识也在不断改变与转型中。之前只是简单地认为细菌体内的生物物质都是无规律、随机分布的,但现在认识到细菌内的染色体分工明细、结构骨架变化不断并且信号传导和生物物质合成有着自己的区域。尽管如此,在光学显微镜下,细菌内的大部分结构都是一团阴影,因此,对于细菌内的分子构成一直处于研究初始状态。这是因为细菌的尺寸远小于光学显微镜的分辨率,因此,无法对它们进行清晰的结构成像。虽然电子显微镜(EM)能够提供很高的成像分辨率,但是该技术还无法应用于细菌成像领域。这是因为在电子显微镜成像过程中,一般需要胶体金免疫标记方法处理细菌。但是该方法需要对细菌进行固化处理,因此会破坏细菌的内结构并影响实验结果。因此电子显微镜无法进行活体成像。
将众多的荧光标记方法与超分辨显微镜结合,就可以解决细菌成像问题。该方法已经被用来研究细菌中众多的蛋白组织结构。例如,Greenfield等[48]用PALM技术研究趋化蛋白焦油受体CheY 和CheW在大肠杆菌中的分布。Biteen等[49]在新月柄杆菌的研究中,使用PALM荧光显微镜给肌动蛋白的类似物MreB的成像,发现该蛋白结构为螺旋型。Ptacin等[50]则用类似方法研究了新月柄杆菌内的分区蛋白(Par)的功能。他们同时发现ATP酶与ParA形成一个狭小、线性的聚体,该聚体穿过细胞体的中间,与真核细胞中的纺锤体功能相似。其中,Fu等[23]成功用PALM荧光显微技术观察了原核细胞的分裂过程,特别是FtsZ蛋白在分裂过程中的分布及功能(图5)。他们清晰地观测到在细胞分裂的过程中,该蛋白在细胞中间形成一个自组装蛋白圆环。该蛋白圆环作为细胞分裂的骨架,对细胞分裂有着重要作用,并为细胞分裂研究提供了新的途径。
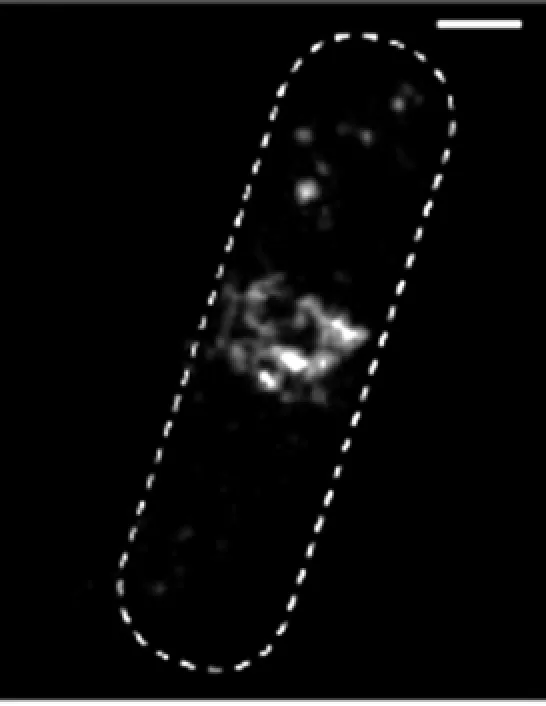
图5 FtsZ蛋白环的PALM成像Fig.5 PALM imaging of Z-ring in E. coli cells.
Wang等[51]用STORM荧光显微技术研究H-NS蛋白在细菌中的分布及其功能(图6)。他们发现H-NS蛋白与DNA结合并在染色体中形成紧凑的聚集体,将与之结合的DNA包裹在其中。由于H-NS可以与E.coli基因组的多个位点相结合,这个形成聚集体的过程可能引起细菌DNA的折叠、收缩,并充当DNA环的锚点。因此,超分辨技术可以进一步帮助研究H-NS蛋白的工作原理及其DNA的三级结构形成的过程。
超分辨荧光显微技术在细菌细胞研究中的应用,展示了该技术在细菌研究中的巨大潜力。考虑到对细菌中染色体和蛋白质结构的研究甚少,生物学家们期待超分辨荧光显微镜在未来几年中,不断推进微生物的发展。
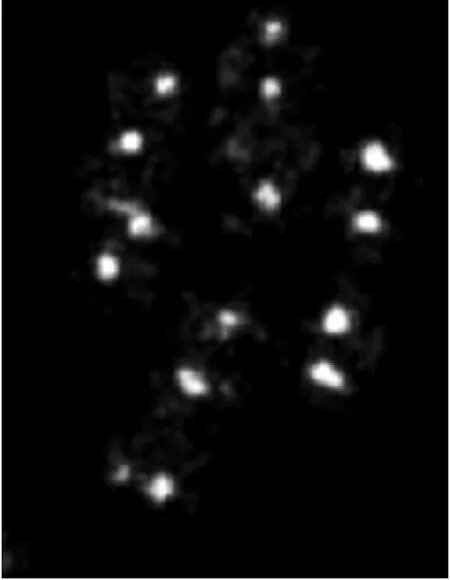
图6 H-NS蛋白在细菌中的成像Fig.6 STORM image of H-NS in E.coli.
2.4超分辨技术在其它领域中的应用
超分辨荧光显微技术不仅可以研究细胞内的结构和作用,也可以对DNA等遗传物质进行单分子成像和研究。由于DNA分子的长度最短仅约50 nm,所以常规的荧光成像方法无法得到清晰的图像。Persson等[17]利用STED高分辨率的优势,成功地得到了单个DNA分子高质量的荧光图像并观测到单个DNA分子中的构想变化(图7)。同时,他们也证明了哺乳动物线粒体的内核质粒大小相同。
STED荧光显微技术也被广泛用于病毒研究中。Chojnacki等[52]利用STED荧光显微镜研究了HIV-1(艾滋病病毒)的外壳蛋白(Env)的分布,并发现HIV病毒外壳蛋白(Env)的分布随着其成熟作用而发生结构改变,并最终形成稳定的外壳蛋白(Env)三聚体(图8)。他们清晰地观测到外壳蛋白Env在胞内Gag水解酶的作用下,将随机分布在病毒表面的外壳蛋白(Env)进行重构而形成有序的外壳蛋白(Env)三聚体。在该重构作用下,病毒细胞的表面结构改变从而让其形态成熟,并使其能够侵入靶标蛋白CD4并繁殖。这些结果说明在病毒表面形成外壳蛋白Env在三聚体HIV-1的成熟的重要标志。这些结果不仅进一步解析HIV-1成熟的机制,也为今后消灭AIDS疾病提供了新的理论依据。
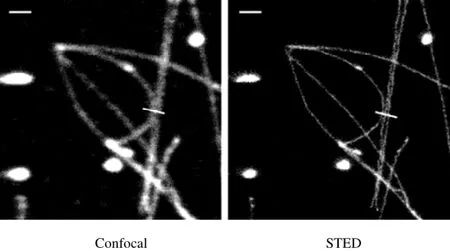
图7 DNA成像技术Fig.7 Confocal and corresponding STED image of DNA.
3 结论及展望
生物技术和纳米技术是21世纪最引人注目的研究领域,而在生命科学与纳米技术走向融合的今天,研究者更关注细胞内纳米尺度分子的动态过程和结构特征,超分辨荧光显微技术应运而生,并在近20年发展飞速,激励大批的科研工作者投身这一研究领域,并作出卓越的贡献。
超分辨技术在生物技术应用过程中存在很多难题,如生物样品的光毒性、活细胞快速成像的分辨率远低于固定样本、成像速度慢等。为了解决这些难题,近年超分辨技术的研究热点集中于提高活细胞成像的时空分辨率。研究者继续研发出更稳定、效率更高、颜色多样的荧光染料或荧光蛋白。并尝试结合不同成像方法的优点,旨在进一步提高精度和准确性。随着超分辨技术的迅猛发展,生物研究者对生物有机体内生化反应过程进行实时动态的观察将成为现实,对人们更加直观的理解生命现象,揭示各种疾病的发病机理,探索新型药物具有重要的意义。
1 Hell S W. Far-field optical nanoscopy[J]. Science, 2007, 316(5828): 1153–1158
2 Deng S, Chen J, Huang Q, et al. Saturated forster resonance energy transfer microscopy with a stimulated emission depletion beam: a pathway toward singlemolecule resolution in far-field bioimaging[J]. Optics Letters, 2010, 35(23): 3862–3864
3 Huang B, Babcock H, Zhuang X. Breaking the diffraction barrier: super-resolution imaging of cells[J]. Cell, 2010, 143(7): 1047–1058
4 Hook R. Micrographia[M]. London: Royal Society of London, 1664
5 Hell S W, Wichmann J. Breaking the diffraction resolution limit by stimulated emission: stimulated-emissiondepletion fluorescence microscopy[J]. Optics Letters, 1994, 19(11): 780–782
6 Bain A J, Marsh R J, Armoogum D A, et al. Timeresolved stimulated emission depletion in two-photon excited states[J]. Biochemical Society Transactions, 2003, 31: 1047–1051
7 Willig K I, Rizzoli S O, Westphal V, et al. STED microscopy reveals that synaptotagmin remains clustered after synaptic vesicle exocytosis[J]. Nature, 2006, 440(7086): 935–939
8 Rankin B R, Kellner R R, Hell S W. Stimulated-emissiondepletion microscopy with a multicolor stimulated-Raman-scattering light source[J]. Optics Letters, 2008, 33(21): 2491–2493
9 Hein B, Willig K I, Hell S W. Stimulated emission depletion (STED) nanoscopy of a fluorescent proteinlabeled organelle inside a living cell[J]. Proceedings of the National Academy of Sciences of the United States of America, 2008, 105(38): 14271–14276
10 Auksorius E, Boruah B R, Dunsby C, et al. Stimulated emission depletion microscopy with a supercontinuum source and fluorescence lifetime imaging[J]. Optics Letters, 2008, 33(2): 113–115
11 Negerl U V, Willig K I, Hein B, et al. Live-cell imaging of dendritic spines by STED microscopy[J]. Proceedings of the National Academy of Sciences, 2008, 105(48): 18982–18987
12 Han K Y, Willig K I, Rittweger E, et al. Threedimensional stimulated emission depletion microscopy of nitrogen-vacancy centers in diamond using continuouswave light[J]. Nano Letters, 2009, 9(9): 3323–3329
13 Ding J B, Takasaki K T, Sabatini B L. Supraresolution imaging in brain slices using stimulated-emission depletion two-photon laser scanning microscopy[J]. Neuron, 2009, 63(4): 429–437
14 Neumann D, Beckers J, Kastrup L, et al. Two-color STED microscopy reveals different degrees of colocalization between hexokinase-I and the three human VDAC isoforms[J]. BMC Biophysics, 2010, 3(1): 4–18
15 Willig K I, Stiel A C, Brakemann T, et al. Dual-label STED nanoscopy of living cells using photochromism[J]. Nano Letters, 2011, 11(9): 3970–3973
16 Wolf T J A, Fischer J, Wegener M, et al. Pump-probe spectroscopy on photoinitiators for stimulated- emissiondepletion optical lithography[J]. Optics Letters, 2011, 36(16): 3188–3190
17 Persson F, Bingen P, Staudt T, et al. Fluorescence nanoscopy of single DNA molecules by using stimulated emission depletion (STED)[J]. Angewandte Chemie-International Edition, 2011, 50(24): 5581–5583
18 Betzig E, Patterson G H, Sougrat R, et al. Imaging intracellular fluorescent proteins at nanometer resolution[J]. Science, 2006, 313(5793): 1642–1645
19 Hess S T, Girirajan T P K, Mason M D. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy[J]. Biophysical Journal, 2006, 91(11): 4258–4272
20 Flors C, Hotta J-I, Uji-I H, et al. A stroboscopic approach for fast photoactivation-localization microscopy with Dronpa mutants[J]. Journal of the American Chemical Society, 2007, 129(45): 13970–13977
21 Hess S T, Gunewardene M. In micro and nano technologies in bioanalysis: methods and protocols[M]. Lee J W, Foote R S. Eds. Totowa: Humana Press, 2009
22 Gould T J, Verkhusha V V, Hess S T. Imaging biological structures with fluorescence photoactivation localization microscopy[J]. Nature Protocols, 2009, 4(3): 291–308
23 Fu G, Huang T, Buss J, et al. In vivo structure of the E. coli FtsZ-ring revealed by photoactivated localization microscopy(PALM)[J]. PLoS One, 2010, 5(9): 12680–12695
24 Matsuda A, Shao L, Boulanger J, et al. Condensed mitotic chromosome structure at nanometer resolution using PALM and EGFP-histones[J]. PLoS One, 2010, 5(9): 12768–12780
25 Hsu C-J, Baumgart T. Spatial association of signaling proteins and F-Actin effects on cluster assembly analyzed via photoactivation localization microscopy in T cells[J]. PLoS One, 2011, 6(8): 23586–23598
26 Mlodzianoski M J, Schreiner J M, Callahan S P, et al. Sample drift correction in 3D fluorescence photoactivetion localization microscopy[J]. Optics Express, 2011, 19(16): 15009–15019
27 Lin H, Centeno S P, Su L, et al. Mapping of surface-enhanced fluorescence on metal nanoparticles using super-resolution photoactivation localization microscopy[J]. Chemphyschem, 2012, 13(4): 973–981
28 Dedecker P, Mo G C H, Dertinger T, et al. Widely accessible method for superresolution fluorescence imaging of living systems[J]. Proceedings of the National Academy of Sciences of the United States of America, 2012, 109(27): 10909–10914
29 Quirin S, Pavani S R P, Piestun R. Optimal 3D single-molecule localization for superresolution microscopy with aberrations and engineered point spread functions[J]. Proceedings of the National Academy of Sciences of the United States of America, 2012, 109(3): 675–679
30 Rust M J, Bates M, Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM)[J]. Nature Methods, 2006, 3(10): 793–795
31 Bates M, Huang B, Dempsey G T, et al. Multicolor super-resolution imaging with photo-switchable fluorescent probes[J]. Science, 2007, 317(5845): 1749–1753
32 Huang B, Jones S A, Brandenburg B, et al. Whole-cell 3D STORM reveals interactions between cellular structures with nanometer-scale resolution[J]. Nature Methods, 2008, 5(12): 1047–1052
33 Vaziri A, Tang J, Shroff H, et al. Multilayer three-dimensional super resolution imaging of thick biological samples[J]. Proceedings of the National Academy of Sciences of the United States of America, 2008, 105(51): 20221–20226
34 Huang B, Wang W, Bates M, et al. Three-dimensional super-resolution imaging by stochastic optical reconstruction microscopy[J]. Science, 2008, 319(5864): 810–813
35 Wu M, Huang B, Graham M, et al. Coupling between clathrin-dependent endocytic budding and F-BAR-dependent tubulation in a cell-free system[J]. Nature Cell Biology, 2010, 12(9): 902–908
36 Burnette D T, Sengupta P, Dai Y, et al. Bleaching/blinking assisted localization microscopy for superresolution imaging using standard fluorescent molecules[J]. Proceedings of the National Academy of Sciences of the United States of America, 2011, 108(52): 21081–21086
37 Jones S A, Shim S-H, He J, et al. Fast, three-dimensional super-resolution imaging of live cells[J]. Nature Methods, 2011, 8(6): 499–505
38 Xu K, Babcock H P, Zhuang X. Dual-objective STORM reveals three-dimensional filament organization in the actin cytoskeleton[J]. Nature Methods, 2012, 9(2): 185–188
39 Veatch S L, Machta B B, Shelby S A, et al. Correlation functions quantify super-resolution images and estimate apparent clustering due to over-counting[J]. PLoS One, 2012, 7(2): 31457
40 Bakshi S, Siryaporn A, Goulian M, et al. Superresolution imaging of ribosomes and RNA polymerase in live Escherichia coli cells[J]. Molecular Microbiology, 2012, 85(1): 21–38
41 English B P, Hauryliuk V, Sanamrad A, et al. Single-molecule investigations of the stringent response machinery in living bacterial cells[J]. Proceedings of the National Academy of Sciences, 2011, 108(31): 365–373
42 Sherman E, Barr V, Manley S, et al. Functional nanoscale organization of signaling molecules downstream of the T cell antigen receptor[J]. Immunity, 2011, 35(5): 705–720
43 Meyer A C, Frank T, Khimich D, et al. Tuning of synapse number, structure and function in the cochlea[J]. Nature Neuroscience, 2009, 12(4): 444–453
44 Van Den Bogaart G, Meyenberg K, Risselada H J, et al. Membrane protein sequestering by ionic protein-lipid interactions[J]. Nature, 2011, 479(7374): 552–555
45 Hell S W. Microscopy and its focal switch[J]. Nature Methods, 2008, 6(1): 24–32
46 Dani A, Huang B, Bergan J, et al. Super-resolution imaging of chemical synapses in the brain[J]. Neuron, 2010, 68(5): 843–856
47 Xu K, Zhong G, Zhuang X. Actin, spectrin, and associated proteins form a periodic cytoskeletal structure in axons[J]. Science, 2013, 339(6118): 452–456
48 Greenfield D, Mcevoy A L, Shroff H, et al. Self-organization of the Escherichia coli chemotaxis network imaged with super-resolution light microscopy[J]. PLoS Biology, 2009, 7(6): 1000137–1000147
49 Biteen J S, Thompson M A, Tselentis N K, et al. Super-resolution imaging in live caulobacter crescentus cells using photoswitchable EYFP[J]. Nature Methods, 2008, 5(11): 947–949
50 Ptacin J L, Lee S F, Garner E C, et al. A spindle-like apparatus guides bacterial chromosome segregation[J]. Nature Cell Biology, 2010, 12(8): 791–798
51 Wang W, Li G W, Chen C, et al. Chromosome organization by a nucleoid-associated protein in live bacteria[J]. Science, 2011, 333(6048): 1445–1449
52 Chojnacki J, Staudt T, Bingen P, et al. Maturationdependent HIV-1 surface protein redistribution revealed by fluorescence nanoscopy[J]. Science, 2012, 338(6106): 524–528
CLCTL99
Application of super-resolution optical microscopy in biology
MAO Xiuhai DU Jiancong HUANG Qing FAN Chunhai DENG Suhui
(Shanghai Institute of Applied Physics,Chinese Academy of Sciences,Jiading Campus, Shanghai 201800,China)
Background:A noninvasive, real-time far-field optical microscopy is needed to study the dynamic function inside cells and proteins. However, the resolution limit of traditional optical microscope is about 200 nm due to the diffraction limit of light. So, it’s hard to directly observe the subcellular structures. Over the past several years of microscopy development, the diffraction limit of fluorescence microscopy has been overcome and its resolution limit is about tens of nanometers. Methods: To overcome the diffraction limit of light, many super-resolution fluoresce microcopies, including stimulated emission of depletion microscopy (STED), photoactivation localization microscopy (PALM) and stochastic optical reconstruction microscopy (STORM), have been developed. Conclusions: These methods have been applied in cell biology, microbiology and neurobiology, and the technology of super-resolution provides a new insight into the life science.
Optical microscopy, Super-resolution, Biology, Nano, Protein
TL99
10.11889/j.0253-3219.2013.hjs.36.060502
毛秀海,男,1986年出生,2008年毕业于华中科技大学,现为上海应用物理研究所博士研究生,从事纳米科学的研究
邓素辉,E-mail: dengsuhui@sinap.ac.cn
2013-04-03,
2013-04-20
