脱氧核酶传感器研究进展
2013-02-13聂绩周颖琳张新祥
聂绩 周颖琳张新祥
(北京大学化学与分子工程学院 北京100871)
1982年,Cech等[1]发现四膜虫核糖体RNA基因转录产物的I型内含子剪切与外显子拼接过程无需任何蛋白质存在,这打破了酶的化学本质是蛋白质的概念,证明了RNA具有酶的催化功能。1983年,Altman等[2]在研究核糖核酸酶P时发现具有切割核糖体RNA前体功能的RNA,并证明其具有全酶活性。因为核酶(ribozyme)的发现,Cech和Altman共享了1989年诺贝尔化学奖。1994年,Breaker和Joyce等[3]利用体外定向分子进化技术筛选得到第一条人工合成的具有切割磷酸二酯键功能的单链DNA,这种具有酶催化功能的DNA就叫做脱氧核酶(deoxyribozyme或DNAzyme)。核酶与脱氧核酶构成了功能核酸的重要部分,核酶包括天然与人工合成核酶;但至少目前为止,自然界没有发现天然脱氧核酶。人工合成的脱氧核酶迄今已有100多种[4],由于其结构与功能的多样性,广泛应用于基因治疗、核酸分析、传感器构建等领域。
1 脱氧核酶基本概念
脱氧核酶的化学本质是DNA。相较于RNA而言,脱氧核酶缺少呋喃糖环2-OH,这决定了脱氧核酶的结构稳定性强于以RNA成构的核酶。所有脱氧核酶均为单链,同RNA相似,可以形成复杂多样的折叠结构并显示出催化功能。脱氧核酶可以催化多类反应,如RNA或DNA切割、DNA连接、DNA水解等[5]。与传统的蛋白酶相比,脱氧核酶具有稳定性高,相对分子质量小,易化学合成、修饰与复制,受pH等环境因素影响小等优势。脱氧核酶非常容易进行以核酸为识别模式或者信号表达模式的传感器构建;另外,脱氧核酶具有对DNA进行连接与断裂的催化能力,可以应用到基因调控与治疗中。
各种功能多样性的脱氧核酶可通过合理设计不同的体外筛选方式获得。筛选过程往往是从一个固相化学合成的DNA随机文库出发,通过合理设计,将能够催化与不具备催化能力的寡核苷酸序列区分开;再利用聚合酶链式反应(PCR),对具有催化能力的序列进行扩增,扩增后获得一个新的文库并应用于下一轮筛选。文库中特异性序列纯度随循环筛选过程进行而增加,直至达到较高催化活力水平时结束筛选。完成筛选后,需要对获得的序列进行表征以及效应评价。一般来说,在体外筛选过程中,需要进行反向筛选,以有效消除没有催化活性的“寄生序列”;也可结合突变与修饰辅助重新筛选提高酶活性。也就是说,应针对不同的筛选目的,合理设计与优化筛选方式。
脱氧核酶的催化功能特性改变了核酸仅仅是编码以及遗传信息携带者的观念。目前,在分子生物学以及基因工程上,脱氧核酶主要是作为一种工具酶存在[6]。脱氧核酶可以在细胞水平上敲除某特异性基因,通过观察该敲除基因对于细胞生理过程中的作用,可以探明其功能。此外,利用脱氧核酶能特异性切断信使RNA,可建立信使RNA水平上有效的基因灭活。通过调控蛋白质表达阻断基因表达,可实现对于某些生理、代谢途径的调节调制。例如,10-23脱氧核酶被用于切断人c-mycmRNA的起始密码子A·UG位点[7]。本文将重点介绍脱氧核酶在传感器领域的应用,其中主要涉及RNA切割型和G-四聚体型。
2 基于脱氧核酶的传感器
传感器是一种对物理或化学信息响应并产生可检测信号的设备。其包括靶标识别与信号转换两个部分:靶标识别元件的作用是识别靶标,必须具备响应迅速、动态范围宽、特异性高、稳定性好以及普适性高的特点;信号转换元件则是将识别事件转换为信号,且信号与靶标浓度之间具有一定的定量关系。通常信号转换元件可以将靶标识别转化为荧光、比色、发光、电化学信号等。功能核酸包括核酶、脱氧核酶、核酸适体以及适体核酶,其靶标范围非常广泛,涉及金属离子、有机小分子、生物大分子甚至细胞与病毒等。正由于可以特异性识别大量靶标,功能核酸已经成为构建靶标识别元件的重要平台[8]。
脱氧核酶在生物基质中比核酶更稳定,且一般尺寸更小,可控性更强,价格便宜且易于合成。因此,目前较广泛研究的是脱氧核酶构建的传感器。构建方式主要是利用靶分子作为辅因子能够促进脱氧核酶切断RNA的反应,从而实现对靶分子(金属离子或者氨基酸等)的定量检测。另一方面,具有过氧化物酶活性的脱氧核酶可以用作识别元件本身或者构建信号转导元件。下面对这两类脱氧核酶传感器作详细阐述。
2.1 RNA切割型脱氧核酶传感器
具有RNA切割功能的脱氧核酶往往需要金属离子或氨基酸等辅因子参与,但也存在无需任何辅因子的RNA切断。10-23型脱氧核酶和8-17型脱氧核酶是两种典型的RNA切割型脱氧核酶,可分别切割嘌呤-嘧啶连接以及AG连接。金属离子的存在可促进DNA的正确折叠以及中间过渡态的稳定,形成催化中心并具有催化活性。此外,还可以屏蔽寡核苷酸的负电荷,以利于酶链与底物链的趋近与结合。目前已发现Pb2+、Mg2+、Zn2+、Mn2+等离子依赖的RNA切割型脱氧核酶。如图1所示为8-17E Pb2+特异性切割脱氧核酶,Pb2+存在下,可在底物链rA位点进行有效切割。目前利用RNA切割型脱氧核酶构建的传感器主要用于检测辅因子,根据信号转导方式不同主要分为荧光型、比色型以及电化学型。
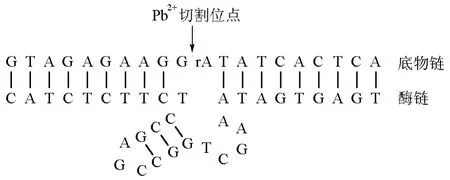
图1 8-17E Pb2+特异性切割脱氧核酶结构示意图
2.1.1 荧光型
分子信标(molecular beacon)是利用发卡结构与靶分子相互作用,使末端标记的荧光基团(F)与猝灭基团(Q)由靠近变为相互远离,从而使荧光信号增强(图2(A))。脱氧核酶切割反应过程类似于分子信标的原理,被称为催化信标。在脱氧核酶催化信标中,可以通过灵活地选取合适的标记位点(图2(B~G))设计不同类型的标记型荧光传感器。Lu等[9]在底物链与酶链末端分别标记荧光基团与猝灭基团(图2(C)),利用底物链断裂导致的荧光增强,可实现Pb2+1×10-8~4×10-6mol/L的定量检测。由于温度升高会造成杂交双链解离,导致荧光背景增加,因此该实验选择在4℃操作。Lu等[10]在以上工作基础上,在底物链的另一端标记猝灭基团(图2(D)),使得即使发生双链解离也不会有荧光背景信号的显著提升。该方法借助分子内及分子间猝灭,可有效降低背景,且不受检测温度的限制。该模型也被成功用于检测、Cu2+[11-12]。为了获得更好的猝灭效率,也有将猝灭基团与荧光基团标记在切割位点附近的方式(图2(E~F))[13-14]。由于靠近切割位点的基团标记会影响催化过程,所以在构建该类型荧光传感器模型时需要进行系统优化[15]。若将脱氧核酶一端固定在固相载体上(图2(G)),通过洗涤可有效降低背景,Pb2+检测限可以达到1×10-9mol/L,比均相溶液降低了一个数量级;同时,固定化酶链有利于传感器的再生与重复使用[16]。

图2 标记型荧光脱氧核酶传感器示意图[8]
一些先进的碳材料也被用于脱氧核酶荧光传感器的构建。Dordick等[17]将脱氧核酶与多壁碳纳米管进行连接,获得了高效稳定的复合物。在底物链一端标记荧光基团可进行脱氧核酶催化能力的评估。Zhang等[18]利用石墨烯与单链DNA具有高亲和作用且能够猝灭荧光素的荧光,构建了基于石墨烯-脱氧核酶的荧光增强型传感器以检测Pb2+,检测限达3×10-10mol/L。
相对于标记型荧光传感器而言,非标记型的普适性更强,不会因为基团标记影响酶催化切割过程。脱氧核酶的无碱基位点能结合外源荧光基团2-氨基-5,6,7-三甲基-1,8-萘啶(ATMND),同时猝灭其荧光。在Pb2+存在下,脱氧核酶对底物链产生切割作用,释放出ATMND,使荧光恢复。Lu等[19]通过引入无碱基位点,建立了一种新型的无标记检测Pb2+的方法,检测限达到4×10-9mol/L。Wang等[20]利用嵌入式荧光染料picogreen(PG)与双链DNA结合后荧光大大增强的性质,构建了非标记检测Pb2+的方法。该方法简单、免修饰、成本低,检测限达1×10-8mol/L(图3)。具有光学性质的高分子材料也被应用到传感器的构建中,Hu等[21]利用水溶性阳离子聚乙烯(PT)与单双链DNA结合表现出不同的荧光性质,设计非标记Pb2+切割型脱氧核酶传感器,检出限达到1×10-8mol/L。
2.1.2 比色型
比色型传感器是利用颜色变化来传导信号。核酸本身没有颜色信号,所以构建比色型脱氧核酶传感器必须要引入信号探针。金纳米颗粒是一种常用的具有高吸光系数的比色探针,其表面等离子体效应与自身粒径大小及颗粒之间距离有关。处于分散状态的金纳米颗粒团聚后,吸收峰值将会从530nm移至650nm,溶液由红变紫(甚至变蓝)。目前基于金纳米颗粒发展的比色型脱氧核酶传感器主要是利用金纳米颗粒之间通过双链DNA连接以及单双链DNA对金纳米颗粒的稳定效果的差异这两种类型进行。

图3 利用PG嵌入型荧光染料的非标记荧光法示意图[20]
Lu等[22]将5'端标记巯基的具有12个碱基的DNA序列修饰于金纳米颗粒表面。脱氧核酶底物链两段延伸并能够和修饰的DNA序列互补配对,形成杂交。当Pb2+存在时,底物链被切割,导致金纳米颗粒分散,溶液呈红色;当Pb2+不存在时,底物链与酶链和修饰DNA序列之间形成稳定杂交,金纳米颗粒被团聚在一起,溶液呈蓝色。因此,利用比色法可以实现对Pb2+的定量检测。切割位点附近的G·T摇摆键对于脱氧核酶17E的活性非常重要,如果将G·T更换为GC,即得到一个突变的脱氧核酶,命名为17Ec。17Ec在Pb2+辅助下完全不具有RNA切割功能。通过调节17E及17Ec的比例,就可以对Pb2+的检测范围实现动态调控。Lu等[23]对该体系条件进行了摸索与优化。由于所设计的体系需要较长时间才能观察到颜色变化,Lu等[24]采取tail-to-tail连接方式代替head-to-tail模式(图4),用42nm的金纳米颗粒代替13nm金纳米颗粒,通过上述改进可在10min内完成检测。在以上体系中,存在Pb2+时,溶液颜色不发生变化;而没有Pb2+时变为蓝色,属于light-down型传感器。为了设计light-up型传感器(即存在Pb2+时颜色发生变化),Lu等[25]采用tail-to-tail型连接,并添加与切断后序列互补的DNA链,可以实现快速light-up检测。更进一步,Lu等[26]将脱氧核酶左右两侧与底物连接臂部分设计为非对称形式,避免了互补序列DNA链的使用。上述方法也被应用到对UO2+2特异性切割的脱氧核酶中,对放射性离子进行检测[27]。
相较于峨眉、昆仑等其他门派对魔教的敌视,武当对于魔教的态度似乎有些模棱两可。理智上他们觉得魔教行事诡谲,不为道义所容,如张翠山在初识殷素素时就曾因她是天鹰教弟子而抗拒与其交往。但情感上他们却又能无视对方的身份,对所接受的人付出真心,如张翠山带殷素素回武当时,师门也对他们表示祝福,并且尽力维护。
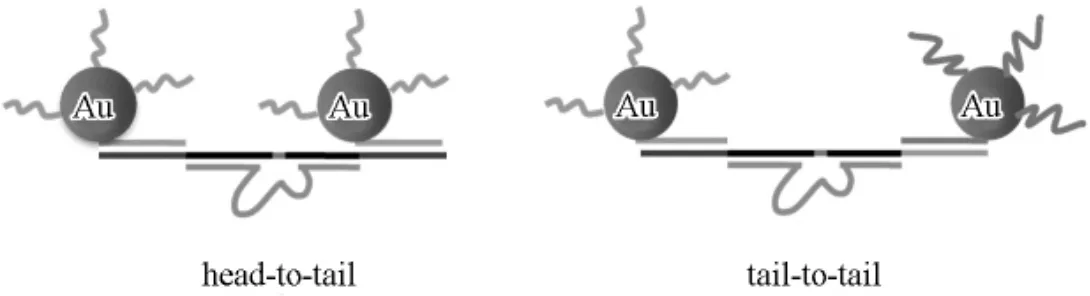
图4 Lu等采用的head-to-tail及tail-to-tail组装方式示意图
单链DNA能够在三羟甲基氨基甲烷与NaCl调节离子强度的环境中稳定金纳米颗粒,阻止红色的金纳米颗粒在高离子强度环境中形成蓝色聚集体。对于Pb2+特异性切割脱氧核酶而言,在Pb2+存在时,底物链被切割为单链DNA,可以稳定金纳米颗粒;无Pb2+时,底物链与酶链之间形成稳定双链杂交,不能稳定金纳米颗粒,溶液变为蓝色聚集体。Lu课题组[28]及Wang课题组[29]基于此原理发展了检测Pb2+的非标记比色型传感器,检出限可低至3×10-9mol/L(图5)。
2.1.3 电化学型
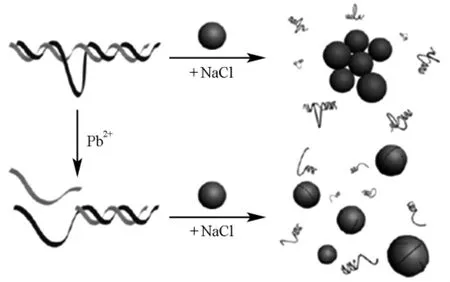
图5 基于金纳米颗粒的非标记Pb2+比色传感器工作原理示意图[28]
与荧光和比色方法相比,电化学方法具有响应迅速、试剂消耗少、灵敏度高等特点。目前发展的电化学型脱氧核酶传感器还比较少。Plaxco等[30]将酶链一端标记亚甲基蓝基团,另一端通过Au—S键固定到金电极表面(图6)。酶链与底物链杂交为双链DNA,使结构刚性增强,亚甲基蓝远离电极,从而使电子传递速率降低。当Pb2+存在时,底物链被切割释放,酶链再度呈单链DNA状态,电子传递速率增强。该方法首次将脱氧核酶修饰于电极表面,未出现酶活性丧失,Pb2+检出限达3×10-7mol/L。Shao等[31]采用带正电荷的六氨合钌与带负电荷的脱氧核酸链骨架静电相互结合作为信号转换元件,DNA-Au生物条形码作信号放大,发展了基于Pb2+特异性切割脱氧核酶的电化学传感器。当Pb2+存在时,被切断的底物链与DNA-Au生物条形码被释放,电极表面六氨合钌减少,其电化学信号降低。由于生物条形码的放大作用,该方法检出限可达1×10-9mol/L。以上方法均具有普适性,可用于其他离子特异性脱氧核酶传感器的构建。同样利用金纳米颗粒,Tian等[32]将脱氧核酶链固定于金纳米颗粒表面,再与底物链杂交。Pb2+切割后,底物链释放,再度呈单链状态的酶链可与固定于电极表面的互补链杂交,利用六氨合钌与脱氧核酸骨架作用获取电信号。该方法极灵敏,检测限可达2.8×10-11mol/L。

图6 Plaxco等在电极表面固定Pb2+特异性切割脱氧核酶进行电化学检测[30]
2.1.4 切割型脱氧核酶作为信号放大元件及构建纳米器件
切割型脱氧核酶不只是被应用于检测辅因子,也作为信号放大或信号转导的工具被应用于其他靶分子传感器的构建中。目前的研究主要是利用Mg2+特异性切割的脱氧核酶,建立了酶放大电化学检测、荧光共振能量转移、比色等方法。
Yu等[33]设计了Mg2+特异性切割10-23型脱氧核酶作为识别探针。此识别探针可与靶序列以及发卡结构的底物链通过杂交作用形成特定构象,在Mg2+作用下,底物链被切断释放,再由该解离的碎片底物链通过杂交捕获于电极表面。由于捕获的碎片底物链一端标记有生物素,可以通过亲和素、生物素之间的特异性亲和作用连接链霉亲和素修饰的磷酸酯酶,即可催化氧化底物产生电化学响应。该方法对于靶标序列的检测限可达5×10-14mol/L。
Willner等借助Mg2+特异性脱氧核酶自催化及分解的脱氧核酶中介过程实现对靶序列DNA的放大检测[34]。此外,他们也利用自组装脱氧核酶线的聚合物,实现对靶序列DNA分析的信号放大[35]。两种放大模式都是建立在切断两端标记猝灭基团与荧光基团的底物链的基础上。前一种模式是在靶序列DNA 1存在时,打开序列3发卡区域,序列1、2、3、4形成稳定复合物。在Mg2+存在下,切断底物链4,获得荧光增强信号(图7)。通过合理的序列设计,可以实现切断与复合物形成的循环放大,检出限可降低3个数量级。在后一种脱氧核酶纳米线的放大过程中,使用两种发卡功能结构。靶序列打开其中一个发卡,激活两种发卡结构的自动化交联过程,这种方式获得的高聚物DNA线中包括Mg2+特异性切割的脱氧核酶结构。利用如图7所示原理,可获得放大的荧光信号,检测限低至10-14mol/L。利用类似的原理,Willner等[36]将标记荧光基团与猝灭基团的底物链替换为能够形成G-四聚体结构的底物链,一旦发生切断解离,释放的序列在氯高血红素(hemin)辅助下具有过氧化物酶催化活性,可以构建比色型核酸适体传感器或者DNA传感器。

图7 Willner等利用Mg2+特异性切割脱氧核酶进行信号放大基本原理图[34]
此外,脱氧核酶也被用以构建可自动行走的DNA纳米器件[37]。将两种不同金属离子Pb2+与Mg2+的脱氧核酶整合在一起,设计出超分子脱氧核酶结构的比色逻辑门[38]。切割型与G-四聚体型脱氧核酶(详见2.2)在功能核酸纳米材料领域占据了重要的地位。
2.2 G-四聚体脱氧核酶传感器
G-四聚体脱氧核酶是一类研究非常热门的脱氧核酶。20世纪90年代末,Sen等[39-40]首次报道了具有过氧化物酶催化活性的脱氧核酶。这种酶与hemin结合能显著增强hemin催化氧化H2O2的能力。G-四聚体结构是由富含G(鸟嘌呤)的脱氧核苷酸序列通过非共价作用形成的特殊高级结构[41]。脱氧核酸碱基之间通常存在氢键相互作用,但这一般是指Waston-Crick碱基互补配对所涉及的A-T、G-C之间的氢键作用。富G序列可以通过鸟嘌呤碱基间的Hoogsteen碱基配对形成更加高级的结构——G-四聚体结构。G-四聚体结构一般可以分为:单分子平行,单分子反平行,双分子反平行,四分子平行(图8)。G-四聚体结构与hemin结合后表现出过氧化物酶催化活性,可催化氧化2,2'-氨基-二(3-乙基-苯并噻唑啉磺酸-6)铵盐或者3,3',5,5'-四甲基联苯胺,应用于比色分析;可催化氧化鲁米诺产生化学发光信号;也可采用电化学方法检测过氧化氢的电催化。G-四聚体-hemin已被当做信号探针或者信号放大模式广泛应用于传感器的构建。
2.2.1 G-四聚体-hemin作为信号报告元件
G-四聚体-hemin已被应用于K+、Hg2+、Ag+、Pb2+、Cu2+等离子的检测。K+具有合适的尺寸,能够进入到G-四聚体结构中,稳定其构型。G-四聚体结构对K+的结合具有一定选择性,在所有碱金属离子中,K+表现出的稳定能力最强,可由形成的具有过氧化物酶催化活性的脱氧核酶进行比色,实现对K+的定量检测[42-43]。Hg2+的检测主要是利用Hg2+能够和两个胸腺嘧啶形成稳定的T-Hg-T结构,通过合理的序列设计,利用T-Hg-T稳定G-四聚体结构[44],或者抑制该结构生成来检测Hg2+[45]。检测Ag+类似于Hg2+,利用Ag+能够与两个胞嘧啶形成C-Ag-C结构,设计该结构使其形成抑制[46]或者稳定[47]具有催化能力的G-四聚体结构,可以进行Ag+定量。Pb2+对G-四聚体结构具有比K+更强的稳定能力,由于Pb2+半径稍小于K+,在Pb2+存在下的G-四聚体结构更加紧凑,可降低与hemin的结合能力。基于上述原理,可以构建荧光型[48]、比色型或化学发光型[49]Pb2+传感器。也有利用金属离子特异性切割脱氧核酶与G-四聚体结构整合,在切割后释放G-四聚体序列,G-四聚体-hemin实现信号输出以检测金属离子。例如Tan等[50]将Cu2+特异性切割脱氧核酶与G-四聚体结构整合,在Cu2+存在时,对发卡结构进行切割释放出G-四聚体序列,再通过与hemin结合,实现比色法检测Cu2+。
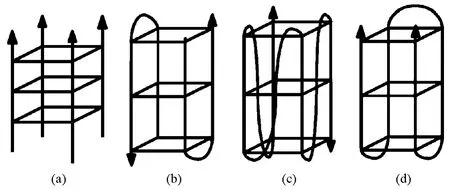
图8 G-四聚体结构
一些核酸适体在与靶分子结合后也能形成G-四聚体结构,与hemin结合可以表现出过氧化物酶催化特性。Dong等[51]利用与凝血酶结合的核酸适体能够在凝血酶、K+以及hemin存在下形成具有催化能力的复合物,进行凝血酶的比色检测。若将hemin更换为一种锌的卟啉化合物Zinc(Ⅱ)-ProtoporphyrinⅨ,形成的G-四聚体复合物可产生荧光信号。三磷酸腺苷的核酸适体也能与靶分子形成G-四聚体结构,利用Zinc(Ⅱ)-ProtoporphyrinⅨ/G-四聚体结构可进行三磷酸腺苷定量检测[52]。
除了检测与靶分子相互作用,G-四聚体-hemin也被作为信息报告元件用于DNA[53]、酶、单碱基多态性[54]等检测,甚至开发出pH敏感的脱氧核酶纳米结构[55]。在作为传感信号报告元件时,往往需要合理的序列设计。在目标事件(例如:与靶序列杂交,酶催化反应等)发生后引入或释放出G-四聚体序列,并在hemin存在条件下折叠为具有催化活力的模拟酶结构,产生信号。目前,基于G-四聚体-hemin作为信号表达测定端粒酶[52-53]、甲基转移酶[56]、限制性内切酶[57]等的方法均有报道。这些传感器的构建将有利于研究酶的生理调制、抑制与激活作用。
2.2.2 G-四聚体-hemin结构与核酸适体/脱氧核酶整合
利用G-四聚体-hemin作为信号报告元件与核酸适体或者脱氧核酶进行结合,拓宽了可检测靶分子的类型,丰富了传感器的设计模式。Willner等[58]将核酸适体与G-四聚体序列整合在一条链上,设计出能与其部分互补的序列。在有靶分子存在时,核酸适体与靶分子的相互作用破坏了两条序列之间的结合,释放的G-四聚体序列可与hemin结合折叠成具有过氧化物酶催化作用的G-四聚体-hemin结构。该方法实现了小分子腺苷与大分子溶菌酶的检测,检出限分别为4×10-6mol/L及4×10-13mol/L。以上方法是基于两条链而言,Willner等[59]在后续工作中还发展了只需要一条链的模式。如图9所示,核酸适体与G-四聚体序列被设计为一个稳定的发卡结构,在没有靶分子存在时,G-四聚体序列被几个互补碱基封闭。当靶分子与核酸适体序列部分相互作用并改变构型时,G-四聚体序列被释放出来,可以产生模拟酶信号。Willner等[60]也在电极表面修饰发卡结构或者引入互补链封闭G-四聚体序列,在靶分子存在时打开发卡释放G-四聚体序列,利用G-四聚体-hemin产生的电信号检测了葡萄糖氧化酶活性、目标DNA和腺苷。在Wang等[61]发展的检测目标DNA以及靶分子的双功能比色探针中,G-四聚体-hemin也被作为信号报告元件使用。两个环结构作为信号识别元件可特异性识别目标DNA与凝血酶,中间使用切断为两截的G-四聚体序列连接,以一种turn-off模式可分别检测两种靶标。

图9 Willner组设计的核酸适体-G-四聚体序列发卡结构工作原理[59]
除了与核酸适体相结合,G-四聚体-hemin也能够与金属离子特异性切割的脱氧核酶相结合,并作为检测金属离子或目标DNA的信号报告元件。Willner等[62]结合Pb2+特异性切割脱氧核酶与G-四聚体-hemin结构发展了灵敏的表面等离子体共振(SPR)以及电化学检测的方法。利用H2O2氧化还原性质进行电化学检测,检测限达1×10-12mol/L;在SPR方法中,利用AuNPs与表面等离子体波电子耦合进行信号放大,检测限可达5×10-15mol/L。Mg2+切割型脱氧核酶也与G-四聚体-hemin结构结合在一起,进行信号表达、放大以及纳米结构的构建(详见2.1.4)。
2.2.3 不同放大模式
G-四聚体-hemin结构具有过氧化物酶活性,可作为信号报告及放大元件,并可结合纳米材料或序列扩增等进行进一步放大,实现超灵敏检测。Ju等[63]使用金纳米颗粒表面修饰G-四聚体序列,加入hemin后形成G-四聚体-hemin功能化的金纳米颗粒。利用该探针标记抗体,再辅助量子点与碳纳米管可进行基于三明治夹心法免疫分析的电致化学发光检测。模拟过氧化物酶功能化的金纳米颗粒还可用于检测目标DNA或者端粒酶活性的信号放大[53]。
滚环扩增是在DNA引物、环状DNA模版以及DNA聚合酶作用下,扩增出含有重复序列单元的单链DNA。利用滚环扩增的方法可以实现对G-四聚体结构的重复扩增,也就是说,使模拟酶数量增加。这相较于G-四聚体-hemin的催化放大而言,放大功能被进一步提升。Mao等[64]利用滚环扩增原位生成若干重复的G-四聚体序列,对目标DNA的串联G-四聚体-hemin信号放大进行比色检测,检测限达1×10-12mol/L。Li等[65]以avidin修饰的磁珠为载体,用核酸适体对靶分子进行特异性识别。再利用滚环扩增延伸串联G-四聚体结构,进行过氧化物模拟酶催化与磁珠双放大。该方法可在2小时内实现对血小板衍生生长因子(PDGF)的检测,检出限为0.2ng/L。滚环扩增的方法虽然可实现超级放大以及超灵敏检测,但其成功依赖于合理的序列设计。
Willner等[66]设计了含有3/4及1/4 G-四聚体模拟酶序列的发卡结构,作为检测目标DNA的功能化信号放大元件。当目标DNA出现时,发卡被打开,引发自动化DNA纳米线的组装。得到的纳米线包含许多G-四聚体结构,可以在结合hemin后进行比色或化学发光的检测。该方法是一种非酶(指蛋白酶)放大模式,检测限可达10-13mol/L。自组装DNA纳米结构的合理设计将对信号放大技术的发展提供更多的可选择模式。
3 总结与展望
脱氧核酶相比于蛋白酶而言稳定性好,相对分子质量小,可控性强、便于与其他功能核酸进行序列设计与整合,既可作为识别元件又可作为信号转导与放大元件,是构建新型生物传感器的有力工具。目前,已经有很多研究小组对于脱氧核酶传感器在分析化学领域的研究做了开创性的工作,但是,进一步拓宽及将其应用于临床或实际检测,还有很多问题需要解决:(1)目前应用于传感器设计的脱氧核酶主要是应用金属特异性切割以及卟啉金属化模拟酶性质的脱氧核酶,而将更多的,具有诸如连接、水解、激酶活性、戴帽活性以及针对于特定靶分子、靶反应的脱氧核酶应用到传感器的构建中,将大大拓展基于脱氧核酶传感器的适用范围与设计灵活性。(2)目前文献报道的许多传感器只是在实验条件下进行概念验证,没有真正应用到实际样品及生物体内。事实上,实际生物样品的复杂基质将会在很大程度上影响传感器的工作。在抗基质干扰方面,还需要做更多的工作,以推进传感器真正应用于临床检测。(3)适体核酶(由含有受体位点的核酸适体与含有催化位点的核酶或脱氧核酶组成)可以作为一种复杂的化学开关,在与靶分子识别作用时变构激活或抑制催化反应。适体核酶的发展与推广为传感器设计、定量检测、高通量筛选以及基因调控提供了新的思路。
[1]Kruger K,Grabowski P J,Zaug A J,et al.Cell,1982,31(1):147
[2]Guerriertakada C,Gardiner K,Marsh T,et al.Cell,1983,35(3):849
[3]Breaker R R,Joyce G F.Chem Biol,1995,2(10):655
[4]Breaker R R.Science,2000,290(5499):2095
[5]Kosman J,Juskowiak B.Anal Chim Acta,2011,707(1):7
[6]Li Y F,Breaker R R.Proc Natl Acad Sci USA,1999,96(6):2746
[7]Sun L Q,Cairns M J,Gerlach W L,et al.J Biol Chem,1999,274(24):17236
[8]Liu J W,Cao Z H,Lu Y.Chem Rev,2009,109(5):1948
[9]Li J,Lu Y.J Am Chem Soc,2000,122(42):10466
[10]Liu J,Lu Y.Anal Chem,2003,75(23):6666
[11]Liu J W,Brown A K,Meng X L,et al.Proc Natl Acad Sci USA,2007,104(7):2056
[12]Liu J,Lu Y.J Am Chem Soc,2007,129(32):9838
[13]Liu Z,Mei S H J,Brennan J D,et al.J Am Chem Soc,2003,125(25):7539
[14]Mei S H J,Liu Z,Brennan J D,et al.J Am Chem Soc,2002,125(2):412
[15]Chiuman W,Li Y F.Nucleic Acids Res,2007,35(2):401
[16]Swearingen C B,Wernette D P,Cropek D M,et al.Anal Chem,2005,77(2):442
[17]Yim T-J,Liu J,Lu Y,et al.J Am Chem Soc,2005,127(35):12200
[18]Zhao X-H,Kong R-M,Zhang X-B,et al.Anal Chem,2011,83(13):5062
[19]Xiang Y,Tong A,Lu Y.J Am Chem Soc,2009,131(42):15352
[20]Zhang L,Han B,Li T,et al.Chem Commun,2011,47(11):3099
[21]Chen X,Guan H L,He Z K,et al.Anal Methods,2012,4(6):1619
[22]Liu J,Lu Y.J Am Chem Soc,2003,125(22):6642
[23]Liu J,Lu Y.Chem Mater,2004,16(17):3231
[24]Liu J,Lu Y.J Am Chem Soc,2004,126(39):12298
[25]Liu J,Lu Y.J Am Chem Soc,2005,127(36):12677
[26]Liu J,Lu Y.Org&Biomol Chem,2006,4(18):3435
[27]Lee J H,Wang Z,Liu J,et al.J Am Chem Soc,2008,130(43):14217
[28]Wang Z D,Lee J H,Lu Y.Adv Mater,2008,20(17):3263
[29]Wei H,Li B L,Li J,et al.Nanotechnology,2008,19(9):1
[30]Xiao Y,Rowe A A,Plaxco K W.J Am Chem Soc,2006,129(2):262
[31]Shen L,Chen Z,Li Y,et al.Anal Chem,2008,80(16):6323
[32]Yang X,Xu J,Tang X,et al.Chem Commun,2010,46(18):3107
[33]Sun C,Zhang L,Jiang J,et al.Biosens Bioelectron,2010,25(11):2483
[34]Wang F A,Elbaz J,Teller C,et al.Angew ChemⅠnt Ed,2011,50(1):295
[35]Wang F,Elbaz J,Orbach R,et al.J Am Chem Soc,2011,133(43):17149
[36]Elbaz J,Moshe M,Shlyahovsky B,et al.Chem Eur J,2009,15(14):3411
[37]Tian Y,He Y,Chen Y,et al.Angew ChemⅠnt Ed,2005,44(28):4355
[38]Bi S,Yan Y,Hao S,et al.Angew ChemⅠnt Ed,2010,49(26):4438
[39]Li Y F,Sen D.Biochem,1997,36(18):5589
[40]Travascio P,Li Y F,Sen D.Chem Biol,1998,5(9):505
[41]Lipps H J,Rhodes D.Trends Cell Biol,2009,19(8):414
[42]Li T,Wang E,Dong S J.Chem Commun,2009(5):580
[43]Yang X,Li T,Li B L,et al.Analyst,2010,135(1):71
[44]Zhang D,Deng M G,Xu L,et al.Chem Eur J,2009,15(33):8117
[45]Li T,Dong S,Wang E.Anal Chem,2009,81(6):2144
[46]Zhou X-H,Kong D-M,Shen H-X.Anal Chem,2009,82(3):789
[47]Li T,Shi L L,Wang E K,et al.Chem Eur J,2009,15(14):3347
[48]Guo L Q,Nie D D,Qiu C Y,et al.Biosens Bioelectron,2012,35(1):123
[49]Li T,Wang E,Dong S.Anal Chem,2010,82(4):1515
[50]Yin B-C,Ye B-C,Tan W,et al.J Am Chem Soc,2009,131(41):14624
[51]Li T,Wang E K,Dong S J.Chem Commun,2008(31):3654
[52]Zhang Z,Sharon E,Freeman R,et al.Anal Chem,2012,84(11):4789
[53]Niazov T,Pavlov V,Xiao Y,et al.Nano Lett,2004,4(9):1683
[54]Wang H Q,Liu W Y,Wu Z,et al.Anal Chem,2011,83(6):1883
[55]Shimron S,Magen N,Elbaz J,et al.Chem Commun,2011,47(31):8787
[56]Li W,Liu Z L,Lin H,et al.Anal Chem,2010,82(5):1935
[57]Zhou Z X,Du Y,Zhang L B,et al.Biosens Bioelectron,2012,34(1):100
[58]Li D,Shlyahovsky B,Elbaz J,et al.J Am Chem Soc,2007,129(18):5804
[59]Teller C,Shimron S,Willner I.Anal Chem,2009,81(21):9114
[60]Pelossof G,Tel-Vered R,Elbaz J,et al.Anal Chem,2010,82(11):4396
[61]Zhang L,Zhu J,Li T,et al.Anal Chem,2011,83(23):8871
[62]Pelossof G,Tel-Vered R,Willner I.Anal Chem,2012,84(8):3703
[63]Lin D,Wu J,Yan F,et al.Anal Chem,2011,83(13):5214
[64]Tian Y,He Y,Mao C D.ChemBioChem,2006,7(12):1862
[65]Tang L,Liu Y,Ali M M,et al.Anal Chem,2012,84(11):4711
[66]Shimron S,Wang F,Orbach R,et al.Anal Chem,2011,84(2):1042
