过渡金属对Mg2Ni氢化物电子结构和热力学稳定性影响:第一性原理研究
2012-12-14陈捷狮蓝志强
陈捷狮,曾 含,王 路,蓝志强,郭 进
(广西大学 物理科学与工程技术学院,南宁 530004)
A2B型 Mg2Ni储氢合金具有储氢量(3.6%,质量分数)大、价格低廉、资源丰富等突出优点,是最具应用前景的储氢合金之一。但是,Mg2Ni合金放氢温度高,反应动力学和热力学性能较差,阻碍了其实际应用[1-2]。近年来,为了提高 Mg2Ni合金的综合性能,国内外学者在实验和理论上进行了大量的研究。如YANG等[3]通过球磨的方法合成了Mg2Ni0.75M0.25合金(XRD相结果显示Mg2Ni0.75M0.25合金为六边形结构),并指出 Mg2Ni0.75M0.25合金经氟化处理后具有良好的储氢性能。吕光烈等[4]研究了以Al和Ti取代Mg2Ni合金中的 Mg原子,发现元素替代后 Mg2Ni形成Mg3MNi2(M=Ti, Al)的立方晶系,且同Mg2Ni相比,Mg3MNi2的吸放氢温度明显降低,循环寿命也得到改善。在理论上,TAKAHASHI等[5]采用DV-Xa方法研究了高温相Mg2NiH4的团簇模型,指出在Mg2Ni合金中Mg—Ni的相互作用占支配地位,且 Ni—H的相互作用明显强于Mg—H的。SETTEN等[6]采用从头算方法研究了过渡金属掺杂对低温相Mg2NiH4的影响,发现Cu或Fe取代一个Ni原子后,氢化物的分解焓大约减少了9.6 kJ/mol,但是Co的替代却增加了氢化物的分解焓。JASEN等[7]采用密度泛函理论方法对低温相Mg2NiH4的电子结构进行了研究,发现Ni—H的相互作用明显大于Mg—H的,且其主要表现为H 1s与Ni 4s3p轨道电子的成键作用。目前,对Mg2Ni合金的研究还在开展中,但是一系列过渡金属分别替代Mg2Ni合金中的部分 Ni原子对合金及其氢化物的晶体结构、电子结构和热力学稳定性的理论研究还较少。研究其电子结构和热力学稳定性为提高Mg2Ni1-xMxH4(M=Mn, Fe, Co, Ni, Cu,x=0.25)的吸放氢动力学性能提供一定的理论指导。密度泛函理论已被广泛应用于材料模拟研究中[8-9],本文作者采用基于密度泛函理论(DFT)的平面波赝势(PW-PP)方法,系统考察了一系列过渡金属M(=Mn, Fe, Co, Cu)分别替代Mg2Ni合金中部分Ni原子对Mg2Ni及其氢化物结构、电子结构和热力学稳定性的影响。
1 计算方法与模型
1.1 计算方法
计算采用基于广义梯度近似(GGA)的密度泛函理论,选用PBE泛函描述电子关联能[10]。从第一性原理出发,将晶体的多电子方程转化为Kohn-Sham方程,选择超软赝势[11](Ultrasoft pseudopotentials)描述电子和离子的相互作用。用CASTEP模块[12]进行结构优化和电子总能计算,平面波截止能取350 eV,简约布里渊区采用Monkhors-Pack方法[13]取适量K点,Mg2Ni及其替代合金K点取为6×6×2,Mg2NiH4及其替代氢化物K点取为4×4×4,M(=Mn, Fe, Co, Ni, Cu)单质的K点取为 12×12×12,H2分子的K点取为3×3×3。结构优化的收敛指标: 总体能量变化小于1.0×10-5eV·atom-1,每个原子的受力小于 0.05 eV·Å,自洽迭代收敛能量为 1.0×10-6eV·atom-1,公差偏移小于0.001 Å,应力偏差小于0.05 GPa。建立a=10 Å简单立方模型来计算H2分子的能量,同时对内坐标进行结构优化。
1.2 晶体结构及模型
Mg2Ni的晶胞模型及在298 K下的Mg2NiH4晶胞模型及替代后的Mg2Ni1-xMxH4(M=Mn, Fe, Co, Ni, Cu,x=0.25)原胞模型如图1所示。
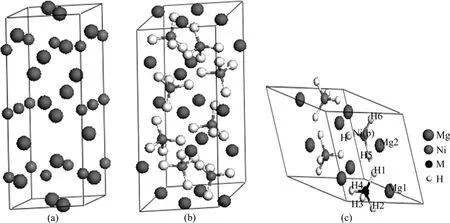
图1 Mg2Ni和Mg2NiH4的晶胞模型以及Mg2Ni0.75M0.25H4的原胞模型Fig.1 Cell models of Mg2Ni (a), Mg2NiH4 (b)and primitive cell model of Mg2Ni0.75M0.25H4(c)
X射线衍射实验表明[14]:Mg2Ni晶体为六方结构,如图1(a)所示,其空间群为P6222(No.180),晶胞参数为a=b=5.205 Å、c=13.236 Å、α=β=90°、γ=120°,Mg原子的空间位置为 6f(0.5, 0, 0.1187)和 6i(0.162,0.324, 0),Ni原子的空间位置为3b(0, 0, 0.5)和3d(0.5,0, 0.5)。计算取Mg12Ni6的单胞,在晶胞中Mg和Ni各有两种独立的位置,分别用Mn、Fe、Co和Cu取代Ni(3d)(取代Ni(3d),Ni(3b)两个位置,计算结果差别很小,且取代Ni(3d)处能量较低),接着,对Mg12Ni6晶胞及替代后 Mg12Ni5M 晶胞进行结构优化。Mg2Ni合金吸氢后首先迅速形成结构不变的 Mg2NiH0.24[15],在298 K下,Mg2Ni合金吸氢后形成Mg2NiH4的单斜结构,如图1(b)所示,其空间群为C2/c(No.15),晶胞参数为a=14.343 Å、b=6.403 8 Å、c=6.483 0 Å、α=γ=90°、β=113.52°,单胞有56个原子,其中,H原子有4种独立的位置,占据着晶格8f位,Mg原子有3种独立的位置分别占据着晶格的8f、4e和4e位,Ni原子只有一种独立的位置占据着晶格的 8f位[16]。考虑Mg2NiH4晶胞特征,本文作者采用其原胞模型进行计算,分别用Mn、Fe、Co和Cu取代 Mg8Ni4H16原胞中的一个Ni原子,如图1(c)所示,原胞由28个原子组成,包含8个Mg原子、3个Ni原子、1个M原子和 16个 H原子,最后,对 Mg8Ni4H16及替代的Mg8Ni3MH16进行结构优化。
2 计算结果与讨论
2.1 结构优化
表1所列为Mg2Ni1-xMx(M=Mn, Fe, Co, Ni, Cu,x=0.25)优化后的晶胞参数。从表中看出,Mg2Ni晶胞参数的计算值与实验测定值很接近,其中,a、b轴的相对误差为 0.36%,c轴的相对误差为 0.15%,说明Mg2Ni所选取的计算条件和参数是合理、可靠的。Mn、Fe、Co和 Cu 替代氢化物中部分 Ni原子后使Mg2Ni晶胞的b和c轴都略有伸长,a轴变短,晶胞体积也发生了明显变化。图2所示为晶胞体积与替代元素的变化关系,替代后Mg2Ni1-xMx晶胞体积的计算值与实验 值[3]吻合较好。
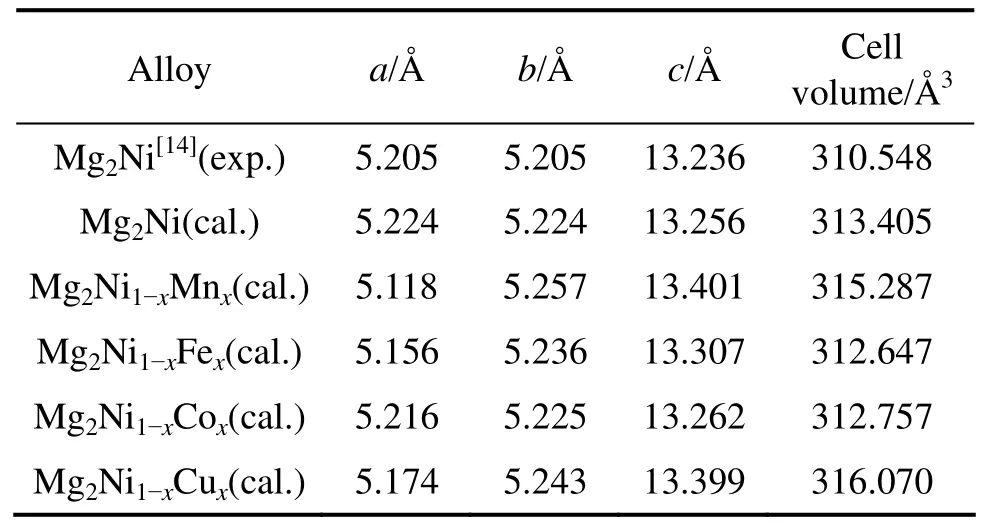
表1 Mg2Ni1-xMx(M=Mn, Fe, Co, Ni, Cu, x=0.25)的晶胞参数Table 1 Cell parameters of Mg2Ni1-xMx(M= Mn, Fe, Co, Ni,Cu, x=0.25)
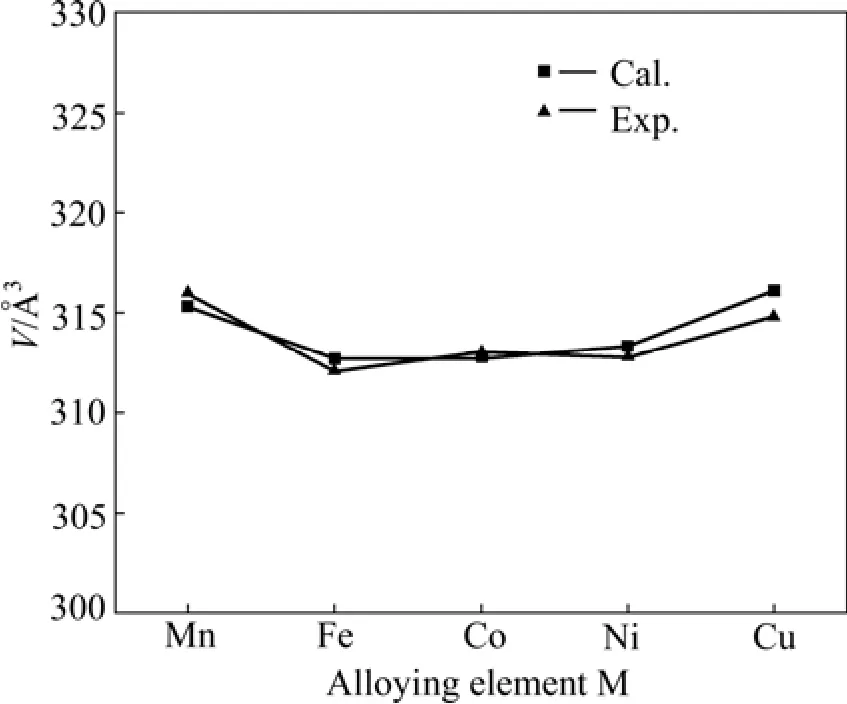
图2 Mg2Ni1-xMx(M=Mn, Fe, Co, Ni, Cu, x=0.25)晶胞体积的计算值和实验值Fig.2 Calculated and experimental unit cell volumes of Mg2Ni1-xMx(M=Mn, Fe, Co, Ni, Cu, x=0.25)
表2列出了Mg2Ni1-xMxH4(M=Mn, Fe, Co, Ni, Cu,x=0.25)结构优化后的晶胞参数。由表2可知,Mg2NiH4晶胞参数与实验值符合很好,其中,a、b轴的相对误差只有0.047%,c轴的相对误差也仅为0.8%。用Mn、 Fe、Co和Cu分别替代Mg2NiH4中部分Ni原子后,Mg2NiH4的晶胞参数发生了明显变化。Mn和Cu的替代导致Mg2NiH4的a轴变长,而Co和Fe的替代使Mg2NiH4的a轴变短,其中,Co的替代引起的a轴变化最明显,大约缩短0.72%。对于b轴,替代后晶胞的b轴都略微增大,其中,Mn替代引起的b轴变化最明显,大约增大2.7%。而对c轴影响最大的是 Cu,Cu的替代使 Mg2NiH4的c轴大约增长15.6%。M(=Mn, Fe, Co, Ni, Cu)替代后Mg2Ni1-xMxH4晶胞体积由大到小的顺序为V(Cu)、V(Ni)、V(Co)、V(Mn)和V(Fe)。
2.2 态密度
图3所示为Mg2Ni1-xMxH4(M=Mn, Fe, Co, Ni, Cu,x=0.25)的总态密度图。图中以费米能级(EF)为能量的参考点。从总态密度(TDOS)图可知,Mn、Fe、Co和Cu分别替代Mg2NiH4中部分Ni原子后的总态密度主峰都下降, 其中,Mn和 Cu的替代下降最明显;同时,替代后的 Mg2Ni1-xMxH4(M=Mn, Fe, Co, Cu,x=0.25)各总态密度的主峰都向深势阱方向移动,其中,Mg2Ni0.75Cu0.25H4的偏移最明显。
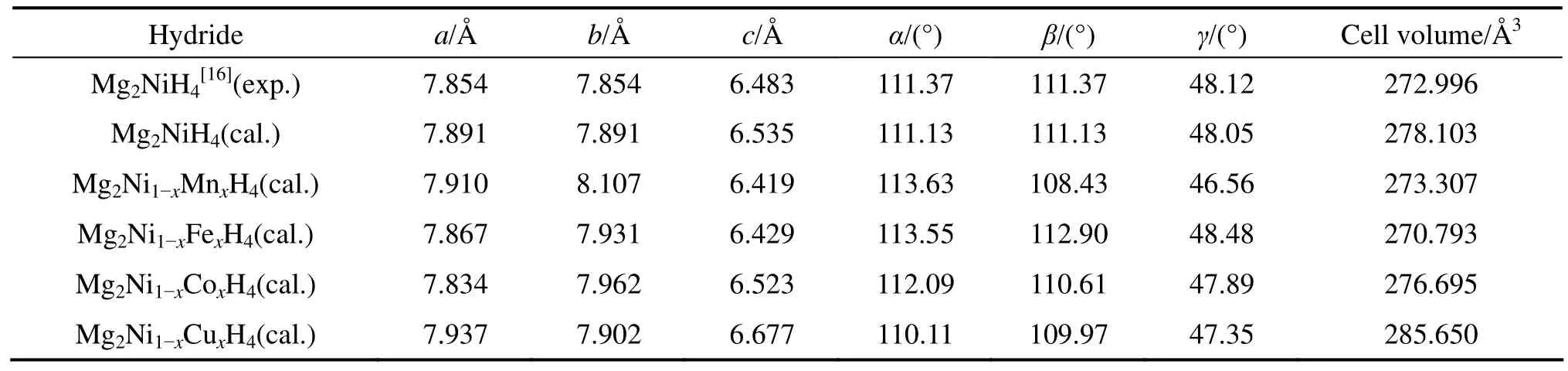
表2 Mg2Ni1-xMxH4(M=Mn, Fe, Co, Ni, Cu, x=0.25)的晶胞参数Table 2 Cell parameters of Mg2Ni1-xMxH4 (M=Mn, Fe, Co, Ni, Cu, x=0.25)

图3 Mg2Ni1-xMxH4(M=Mn, Fe, Co, Ni, Cu, x=0.25)的总态密度图Fig.3 Total density of states (DOSt)of Mg2Ni1-xMxH4 (M=Mn, Fe, Co, Ni, Cu, x=0.25)
由于替代后态密度的整体变化趋势大体相同且Cu变化最明显,所以,进一步研究Cu替代前后总态密度及分波态密度情况。图4所示为 Mg2NiH4和Mg2Ni0.75Cu0.25H4的总态密度及分波态密度。由图4(a)可得,Mg2NiH4的导带和价带间的禁带宽度为1.72 eV,这与BROEDERSZ等[17]的计算值接近,表明 Mg2NiH4具有明显的非金属性。在费米能级(EF)以下,总态密度主要由 3个成键峰构成,其中,位于-8.8~-6.2 eV能量范围内的总态密度峰主要由 Mg 3s、Ni 4s与H 1s轨道的电子贡献;位于-6.2~-3.7 eV能量范围内的总态密度峰主要由Mg 2p,Ni 3p(3d)及H 1s轨道的电子贡献;位于-2.7~0 eV能量范围内的总态密度峰主要由Mg 3s2p、Ni 3p(3d)及H 1s轨道的电子贡献。同时,H原子和Ni原子的态密度在价带部分有较大重叠,而Mg原子和H原子的态密度在价带部分重叠则较小。Cu替代Mg2NiH4中部分Ni原子后的总态密度发生了明显的变化(见图4(b)),其主要特征如下:1)在费米能级(EF)下,比替代前多了一个位于-5.2~-4.8 eV的成键峰,这个成键峰主要由替代元素Cu 3d轨道的电子贡献。2)替代后的总态密度相对替代前总态密度峰值下降很多,表明Cu的替代使Mg、Ni和 H原子参与成键的电子数减少。3)位于-10.5~-7.9 eV能量范围内的总态密度峰主要由Mg 3s、Ni 4s、Cu 4s与H 1s轨道的电子贡献;位于-7.9~-5.2 eV能量范围内的总态密度峰主要由 Mg 2p、Ni 3p3d、Cu 3p3d及H 1s轨道的电子贡献;位于-4.2~-1.8 eV能量范围内的总态密度峰主要由Mg 3s2p、Ni 3p3d、Cu 3p及H 1s轨道的电子贡献。4)在费米能级(EF)以下,Cu原子和H原子的态密度重叠区域比Ni原子和H原子的态密度重叠区域大,说明在Mg2Ni0.75Cu0.25H4中Cu原子和H原子的成键电子数多于Ni原子和H原子的成键电子数。
2.3 电荷等密度和原子间键序
图5所示为Mg2Ni1-xMxH4(M=Mn, Fe, Co, Ni, Cu,x=0.25)的电荷等密度图。其中,图5(a)和(b)所示分别为Mg2NiH4两个不同截面的电荷等密度图;图5(c)和(d)所示分别为 Mg2Ni0.75Cu0.25H4对应于图5(a)和(b)的两个电荷等密度图;图5(e)、(f)和(g)分别表示 Mg2-Ni0.75Mn0.25H4、Mg2Ni0.75Fe0.25H4和 Mg2Ni0.75Co0.25H4对应于图5(a)的电荷等密度图。从图5可知,在氢化物Mg2Ni1-xMxH4中,H原子和Mg原子的电子云重叠不明显,而H原子与Ni、M(M=Mn, Fe, Co, Cu)原子的电子云存在较大的重叠,这说明 H原子和 Ni、M原子具有很强的相互作用。比较图5(a)和(c)的可知,Ni原子和H原子的电子云重叠明显少于Cu原子和H的电子云重叠。比较图5(b)和(d)图可知,Cu的替代使Ni(b)—H的电子云重叠减少,说明Cu的替代会减弱Ni(b)和H原子的相互作用。这与态密度图反映的情况一致。
为了分析Mg2Ni1-xMxH4(M=Mn, Fe, Co, Ni, Cu,x=0.25)的成键作用,采用下列表达式[18]计算氢化物中部分原子单位键长的键序BOs(scale bond order):

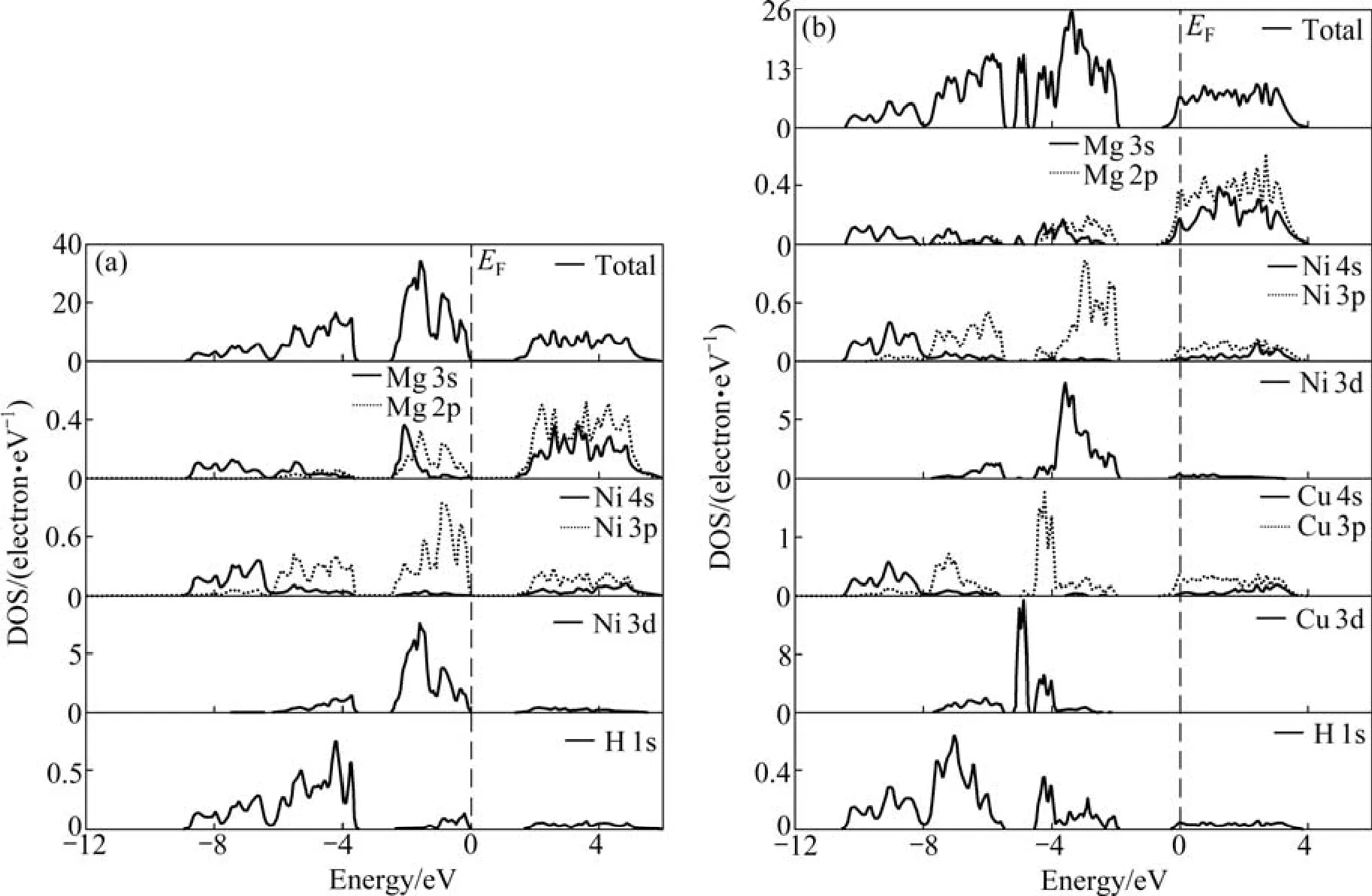
图4 Mg2NiH4和Mg2Ni0.75Cu0.25H4的总态密度及分波态密度Fig.4 Total and partial density of states of Mg2NiH4 (a)and Mg2Ni0.75Cu0.25H4 (b)
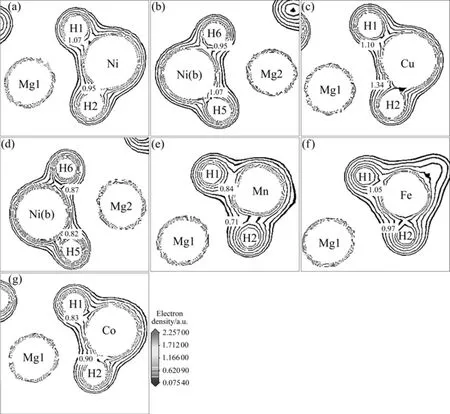
图 5 Mg2Ni1-xMxH4(M=Mn, Fe, Co, Ni, Cu,x=0.25)的电荷等密度图Fig.5 Contour maps of electron density of Mg2NiH4 ((a), (b)), Mg2Ni0.75Cu0.25H4 ((c), (d)),Mg2Ni0.75Mn0.25H4 (e), Mg2Ni0.75Fe0.25H4 (f)and Mg2Ni0.75Co0.25H4 (g)
式中:BO和BL分别表示原子间的电荷布居和键长。键序BOs可用来定性评价结构中两原子相互作用的强弱,若BOs为正且键序值越大,说明两原子是共价键结合且相互作用越强;若键序值为负,说明两原子之是离子键结合[18]。Mg2Ni0.75M0.25H4(M=Mn, Fe, Co, Ni,Cu)键序与替代元素的计算结果如图6所示。图中,M—H表示M与周围所对应4个氢原子(即它周围四面体上的4个氢原子)的平均键序;Ni—H表示氢化物原胞中所有 Ni原子和 H原子的平均键序;Mg2—Ni(b)、Mg1—H2和M—Mg1分别代表单个原子间的键序(具体原子位置见图1(c))。观察键序随M的变化可以发现:Ni—H、Mg2—Ni(b)和Mg1—H2的键序随M变化较小,而M—H和Mg1—M的键序随M变化却很明显,且 M—H键的相互作用越强,对应的Mg1—M 键的相互作用就越弱。TAKAHASHI等[5]通过 DV-Xa法对 Mg2NiH4团簇模型进行研究时也发现了同样的情况。比较各键序大小发现,Ni—H和 M—H的键序始终为正,Mg1—H2的键序为负,且Ni—H和 M—H的键序值较大,这说明氢化物中的Ni—H和M—H键为共价键,Mg1—H2键为离子键,并且 Ni—H和 M—H键存在较强的相互作用。所以,Mg2Ni1-xMxH4放氢困难的一个主要原因Ni—H和M—H键的强相互作用。观察Ni—H的键序随M的变化可知,Mn、Fe和 Co分别替代Mg2NiH4中部分Ni原子后对氢化物中的Ni—H的影响较小,而Cu替代则减弱了Ni—H键的相互作用。
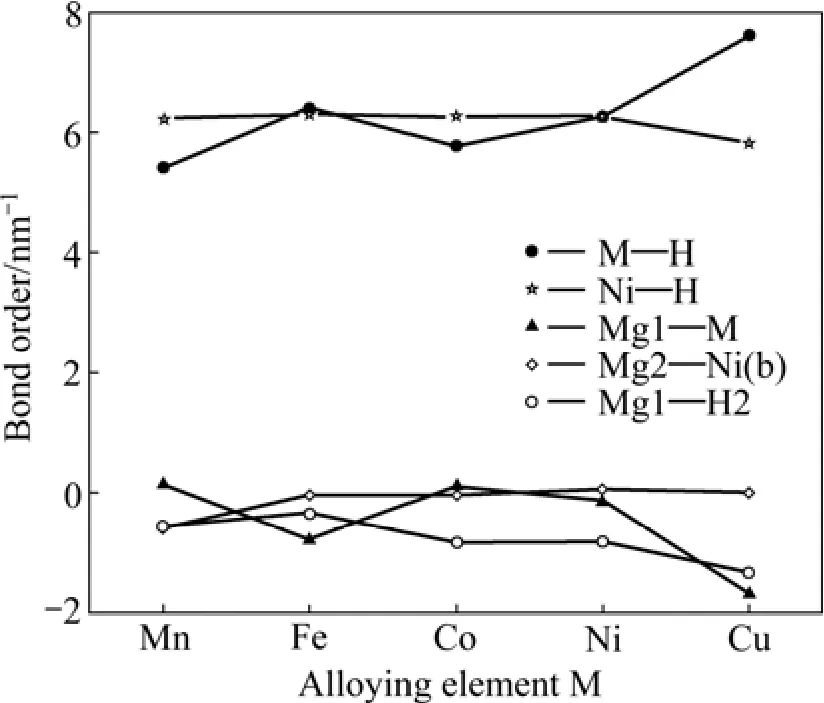
图6 Mg2Ni1-xMxH4(M=Mn, Fe, Co, Ni, Cu, x=0.25)键序与替代元素的变化关系Fig.6 Relationship between bond order and substituting element for Mg2Ni1-xMxH4 (M=Mn, Fe, Co, Ni, Cu, x=0.25)
2.4 热力学稳定性
生成焓ΔH是研究储氢材料热力学稳定性的重要参数,其表达式为生成物的总能量减去反应物的总能量[17]:

若生成焓ΔH的值为负,表明该反应是放热反应;且反应过程放出的热量越多,生成物的结构就越稳定[19]。一般情况下,Mg2Ni合金吸氢过程的反应式如下[20]:

通过式(3)可得Mg2NiH4的生成焓ΔH表达式为
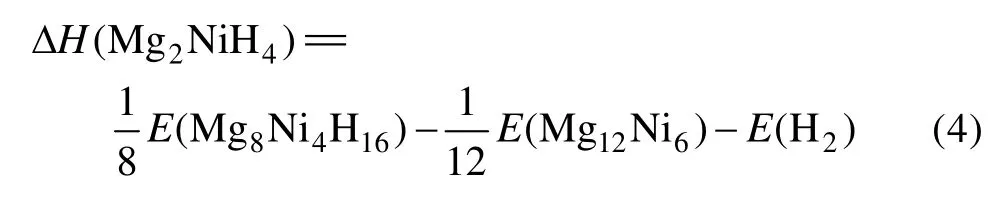
元素替代后,Mg2Ni合金吸氢过程的反应式可表示为[21]
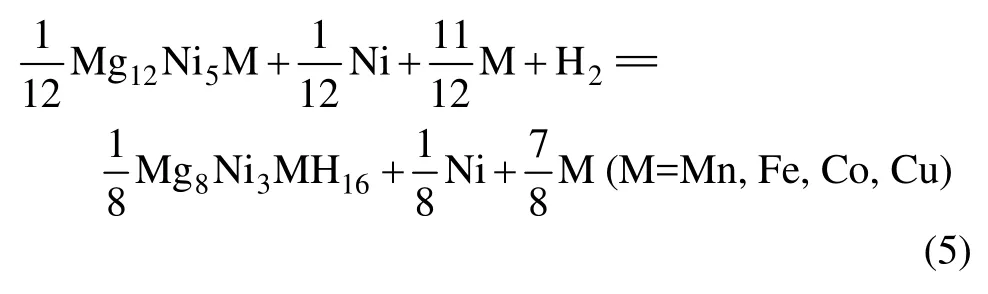
所以,通过下式可得 Mg2Ni1-xMxH4(M=Mn, Fe,Co, Cu,x=0.25)的生成焓ΔH表达式为
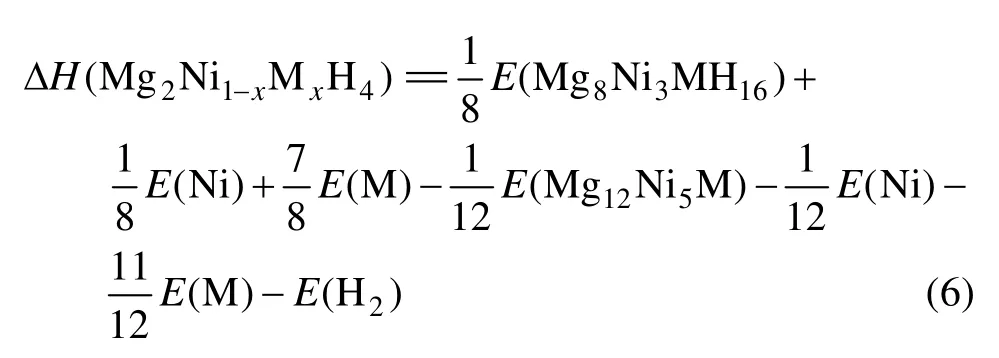
Mg12Ni6及其替代合金,Mg8Ni4H16及其替代氢化物,还有计算过程要用到的Mn、Fe、Co、Ni、Cu单质和H2能量如表3所列。将表3的能量代入式(4)和(6), 可算出Mg2Ni0.75M0.25H4(M=Mn, Fe, Co, Ni, Cu)的生成焓(见表4)。
比较Mg2Ni1-xMxH4(M=Mn, Fe, Co, Ni, Cu,x=0.25)生成焓的计算值和实验值可以发现,生成焓的计算值和实验值[3,22]符合得较好。在所研究的替代元素中,Fe和Co分别替代Mg2NiH4中的部分Ni原子对Mg2NiH4结构稳定性影响较小,而Mn和Cu替代则较好地降低了 Mg2NiH4的结构稳定性,尤其是 Cu的替代使Mg2NiH4结构稳定性下降最明显。TAKAHASHI等[5]也指出,Cu的添加可以很好地降低 Mg2NiH4的结构稳定性。Mg2Ni0.75Cu0.25H4结构稳定性下降的一个原因可能是Cu的替代使氢化物中Ni—H键减弱引起的。
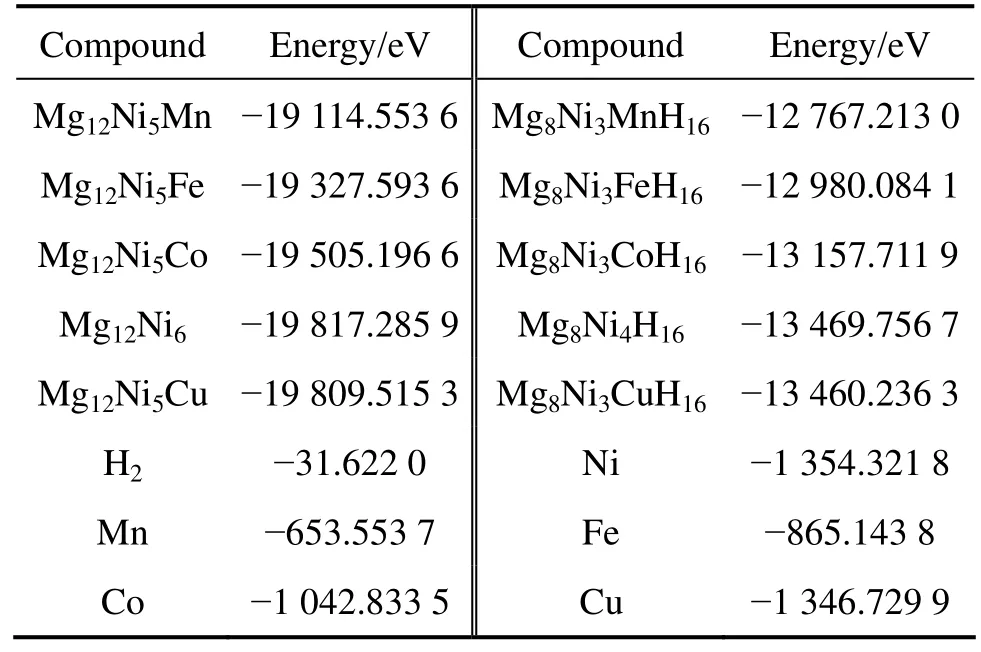
表3 金属、金属间化合物和氢化物的能量计算值Table 3 Calculated energies of metals, intermetallics and hydrides
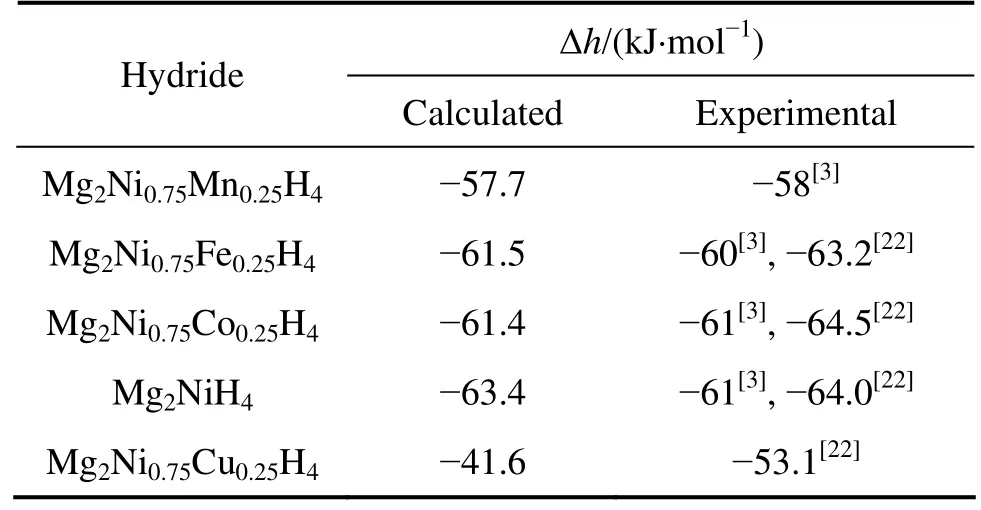
表4 Mg2Ni1-xMxH4(M=Mn, Fe, Co, Ni, Cu, x=0.25)生成焓的计算值和实验值Table 4 Calculated and experimental formation enthalpies of Mg2Ni1-xMxH4 (M=Mn, Fe, Co, Ni, Cu, x=0.25)
2.5 氢化物的解离能
为了考察Mn、Fe、Co和Cu替代Mg2NiH4中的Ni原子前后体系的放氢性能,计算了Mg2Ni0.75M0.25H4晶胞中 Ni(b)原子周围解离出最邻近的两个氢原子(即去除图1(c)中H5和H7原子)所需的能量,计算采用的表达式如下[23]:
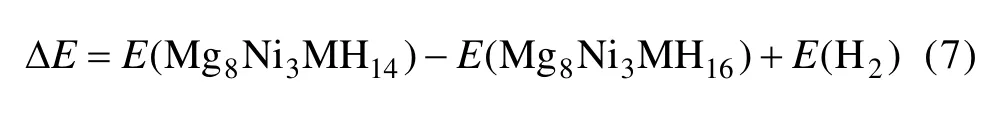
其中:ΔE表示 Mg2Ni0.75M0.25H4晶胞中 Ni(b)原子周围解离出最邻近的两个氢原子所需的能量;E(Mg8Ni3MH16)和E(Mg8Ni3MH14)则分别表示Mg2Ni0.75M0.25H4解离两个氢原子前后的总能量;E(H2)表示氢气分子的能量,其值为-31.622 0 eV,计算结果如表5所列。计算结果表明:Mg2Ni0.75Cu0.25H4的解离能最小,这说明Cu替代Mg2NiH4中部分Ni原子可以提高 Mg2NiH4的放氢性能。SELVAM 等[22]也指出,Cu的添加可以提高Mg2NiH4的放氢性能。
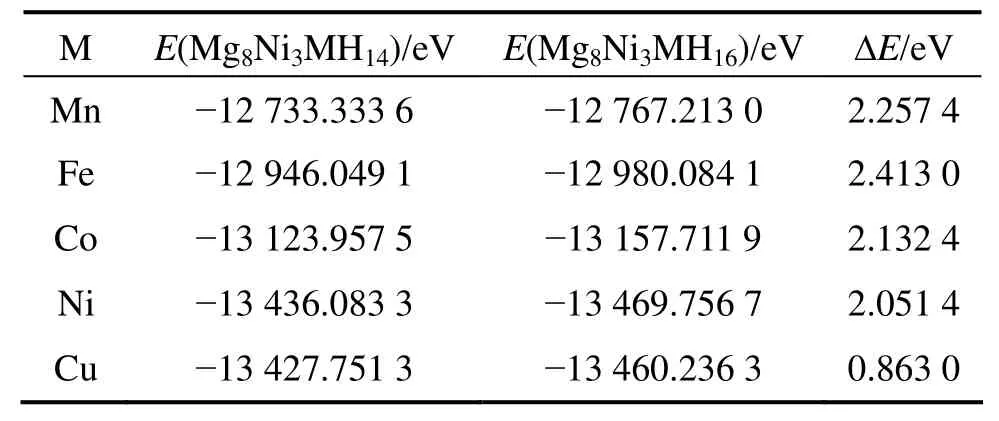
表5 Mg2Ni0.75M0.25H4(M=Mn, Fe, Co, Ni, Cu)氢化物解离能的计算值Table 5 Calculated dissociated energies of Mg2Ni0.75M0.25H4(M=Mn, Fe, Co, Ni, Cu)
3 结论
1)采用基于密度泛函理论(DFT)的平面波赝势(PW-PP)方法对Mg2Ni1-xMx(M=Mn, Fe, Co, Ni, Cu,x=0.25)合金及其对应的氢化物Mg2Ni1-xMxH4(M=Mn, Fe,Co, Ni, Cu,x=0.25)的结构进行优化,计算结果和实验值基本符合。
2)在氢化物Mg2Ni0.75M0.25H4中,Ni—H和M—H键表现为共价键,Mg—H键表现为离子键,且Ni—H和M—H键的相互作用大于Mg—H键的相互作用,这说明 Mg2Ni0.75M0.25H4吸放氢性能主要取决于Ni—H和M—H键的强相互作用。Mn、Fe和Co的部分替代对Ni—H键的相互作用影响较小,而Cu的替代则减弱了Ni—H键的相互作用,这可能是Cu替代后氢化物结构稳定性降低的一个原因。
3)生成焓的计算结果表明:Mg2Ni0.75M0.25H4(M=Mn, Fe, Co, Ni, Cu )的生成焓分别为-57.7、-61.5、-61.4、63.4和41.6 kJ/mol,与实验值符合得较好。
[1]GROCHALA W, EDWARDS P P.Thermal decomposition of the non-interstitial hydrides for the storage and production of hydrogen[J].Chemical Reviews, 2004, 104(3): 1283-1316.
[2]SCHLAPBACH L, ZUTTEL A.Hydrogen-storage materials for mobile applications[J].Nature, 2001, 414: 353-358.
[3]YANG Hua-bin, YUAN Hua-tang, JI Jing-tao, SUN Hua, ZHOU Zuo-xiang, ZHANG Yun-shi.Characteristics of Mg2Ni0.75M0.25(M=Ti, Cr, Mn, Fe, Co, Ni, Cu and Zn)alloys after surface treatment[J].Journal of Alloys and Compounds, 2002, 330:640-644.
[4]吕光烈, 陈林深, 胡秀荣, 王连邦, 袁华堂.Mg3MNi2(M=Ti,Al)的晶体结构[J].金属学报, 2001, 37(5): 459-462.LÜ Guang-lei, CHEN Lin-shen, HU Xiu-rong, WANG Lian-bang, YUAN Hua-tang.The crystal structure of new hydrogen storage Mg3MNi2(M=Ti, Al)alloy[J].Acta Metallurgica Sinica, 2001, 37(5): 459-462.
[5]TAKAHASHI Y, YUKAWA H, MORINAGA M.Alloying effects on the electronic structure of Mg2Ni intermetallic hydride[J].Journal of Alloys and Compounds, 1996, 242(1/2):98-107.
[6]van SETTEN M J, de WIJS G A, BROCKS G.Ab initio study of the effects of transition metal doping of Mg2NiH4[J].Physical Review B, 2007, 76(7): 75125-75133.
[7]JASEN P V, GONZALEZ E Z, BRIZUELA G, NAGEL O A,GONZALEZ G A, JUAN A.A theoretical study of the electronic structure and bonding of the monoclinic phase of Mg2NiH4[J].International Journal of Hydrogen Energy, 2007, 32(18):4943-4948.
[8]罗湖斌, 胡青苗, 杨 锐.合金化对β钛合金热膨胀系数的影响: 第一性原理研究[J].中国有色金属学报, 2010, 20(s1):399-403.LOU Hu-bin, HU Qing-miao, YANG Rui.Effect of alloying on thermal expansion efficient ofβtitanium alloy: First principles study[J].The Chinese Journal of Nonferrous Metals, 2010,20(s1): 399-403.
[9]刘奕新, 梁 初, 蒋 龙, 黎光旭, 韦文楼, 郭 进.Li-Al-N-H 系络合物贮氢反应的第一性原理研究[J].中国有色金属学报, 2009, 19(1): 108-113.LIU Yi-xin, LIANG Chu, JIANG Long, LI Guang-xu, WEI Wen-lou, GUO Jin.Investigations on Li-Al-N-H complex for hydrogen storage by first principle[J].The Chinese Journal of Nonferrous Metals, 2009, 19(1): 108-113.
[10]MARLO M, MILMAN V.Density-functional study of bulk and surface properties of titanium nitride using different exchange-correlation functionals[J].Physical Review B, 2000,62(4): 2899-2907.
[11]VANDERBILT D.Soft self-consistent pseudopotentials in a generalized eigenvalue formalism[J].Physical Review B, 1990,41(11): 7892-7895.
[12]SEGALL M D, LINDAN PHILIP J D, PROBERT M J,PICKARD C J, HASNIP P J, CLARK S J, PAYNE M C.First-principles simulation: Ideas, illustrations and the CASTEP code[J].Journal of Physics: Condensed Matter, 2002, 14(11):2717-2744.
[13]MONKHORST H J, PACK J D.Special points for Brillouin-zone integrations[J].Physical Review B, 1976, 13(12):5188-5192.
[14]DARRIET B, SOUBEYROUX J L, PEZAT M.Structural and hydrogen diffusion study in the Mg2Ni-H2system[J].Journal of the Less Common Metals, 1984, 103(1): 153-162.
[15]NOREUS D, WERNER P.Structural studies of hexagonal Mg2NiHx[J].Acta Chemica Scandinavica A, 1982, 36(10):874-851.
[16]ZOLLIKER P, YVON K, JORGENSEN J, ROTELLA F J.Structural studies of the hydrogen storage material magnesium nickel hydride (Mg2NiH4): 2.Monoclinic low-temperature structure[J].Inorganic Chemistry, 1986, 25(20): 3590-3593.
[17]BROEDERSZ C P, GREMAUD R, DAM B, GRIESSEN R.Highly destabilized Mg-Ti-Ni-H system investigated by density functional theory and hydrogenography[J].Physical Review B,2008, 77(2): 24204-24214.
[18]ZHANG R J, WANG Y M, CHEN D M, YANG R, YANG K.First-principles calculations of LaNi4Al-H solid solution and hydrides[J].Acta Materialia, 2006, 54(2): 465-472.
[19]LI S L, WANG P, CHEN W, LUO G, CHEN D M, YANG K.Hydrogen storage properties of LaNi3.8Al1.0M0.2(M=Ni, Cu, Fe,Al, Cr, Mn)alloys[J].Journal of Alloys and Compounds, 2009,485(1/2): 867-871.
[20]REILLY J J, WISWALL R H.The reaction of hydrogen with alloys of magnesium and Nickel and the formation of Mg2NiH4[J].Inorganic Chemistry, 1968, 7(11): 2254-2256.
[21]ZENG Yan-li, FAN Ke, LI Xiao-yan, XU Bao-en, GAO Xiao-zhen, MENG Ling-peng.First-principles studies of the structures and properties of Al- and Ag-substituted Mg2Ni alloys and their hydrides[J].International Journal of Hydrogen Energy,2010, 35(19): 10349-10358.
[22]SELVAM P, VISWANATHAN B, SWAMY C S, SRINIVASAN V.Studies on the thermal characteristics of hydrides of Mg,Mg2Ni, Mg2Cu and Mg2Ni1-xMx(M= Fe, Co, Cu or Zn; 0 [23]LI S, JENA P, AHUJA R.Dehydrogenation mechanism in catalyst-activated MgH2[J].Physical Review B, 2006, 74(13):132106-132109.
