锂离子电池正极材料进展
2012-12-08韩虹羽
明 博 韩虹羽
(浙江凯圣氟化学有限公司,浙江 衢州 324004)
综 述
锂离子电池正极材料进展
明 博 韩虹羽
(浙江凯圣氟化学有限公司,浙江 衢州 324004)
介绍了锂离子电池的发展阶段、工作原理及特点,叙述了锂电池已商业化正极材料钴酸锂、锰酸锂、磷酸铁锂的特性、合成方法及其优缺点,纳米技术锂离子电池正极材料应用及其合成方法。认为应根据现有正极材料出现的问题,通过掺杂、包覆、加入辅助剂和表面修饰改性等方法减低成本,利用纳米材料的优点和微米材料优良的稳定性和容易制备的优点合成纳微分层结构的材料解决纳米材料的低热力学稳定性、团聚及与电解液发生副反应等问题;可以尝试着探索新的方法合成纳米级颗粒,并将最优的方法应用于新材料和经典电极材料的制备,从而充分发挥纳米级材料的尺寸效应和表面效应,改善电极材料的电化学活性,有助于推进纳米正极材料的工业化进程。
锂离子电池;电池正极材料;钴酸锂;锰酸锂;磷酸铁锂;纳米材料;进展
1990年日本Sony公司研制出锂离子电池并于1991年实现商品化,在随后的10余年中,其商业化进程取得了突飞猛进的发展。作为新能源电池,锂离子电池因其卓越的高性能价格比,其应用前景被十分看好。目前为止,锂离子电池不但已经在笔记本、移动电话、摄录机、武器装备等移动电子终端设备领域占据了主导地位,而且在国内外正竞相开发的电动汽车、航天和储能等领域也崭露头角,尤其是大容量锂离子二次电池成为研究的热点[1-3]。
锂离子电池的发展经过了锂电池和锂离子电池阶段。锂电池一般指锂一次电池和锂二次电池,这种电池的负极是金属锂,正极用 MnO2、SnCl2、SO2、(CFx)n等材料。其中,锂一次电池的研究始于20世纪50年代,20世纪70年代进入实用化;锂二次电池是以金属锂作为电池负极的二次电池体系,在反复充放电过程中锂离子易沉积在金属锂负极的表面形成锂枝晶,当枝晶发展到一定程度时,一方面会发生折断,产生“死锂”,造成锂的不可逆;另一方面更严重的是,枝晶穿过隔膜,将正极与负极连接起来,结果产生大电流,生成大量的热,使电池着火,甚至发生爆炸,从而引起严重的安全问题[2]。
由于锂二次电池存在无法克服的缺点,因此锂二次电池最终被锂离子电池取代。1987年,Auborn等第1次装配了以MnO2或WO2为负极,LiCoO2为正极的摇椅式电池。与金属锂为负极的锂二次电池相比,这些电池的安全性和循环性能大大提高。但由于负极材料(LiMnO2、LiWO2等)的嵌锂电位较高(0.7~2.0 V,Li/Li+),因此未能实际应用。
1990年日本Sony能源技术公司首先推出LixC6l LiClO4-PC+EClLi1-xCO2实用型锂离子电池。该电池既克服了锂二次电池循环寿命低、安全性差的缺点,又较好地保持了锂二次电池高电压、高比能量的优点。由此,二次锂离子电池在全世界范围内掀起了研究开发热潮,并取得了较大的进展[4]。在国内锂离子二次电池开发也将是我国新能源汽车产业发展的关键。
2012年4月国务院讨论通过节能与新能源汽车产业发展规划,明确了今后电动汽车的发展目标,争取到2015年,纯电动汽车和插电式混合动力汽车累计产销量达到50万辆,到2020年超过500万辆。而锂离子电池成为车用动力电池的主要发展方向,2012年4月科技部发布的《电动汽车科技发展“十二五”专项规划》指出“以锂离子动力电池为重点的车用电池产业竞争能力等方面处于国际先进水平”。由此,可以预见锂离子电池将是我国新能源战略的一个重要组成部分。对锂离子电池的发展现状和前景的研究具有必要性。
1 锂离子电池原理及特点
1.1 定义及工作原理
锂离子电池主要由正极、负极、能传导锂离子的电解液以及把正负极隔开的隔离膜构成。锂离子电池负极是碳素材料,如石墨等。正极则是含锂的过渡金属氧化物,如LiCoO2、LiMn2O4等。在充电时正极材料中的锂离子开始脱离正极透过隔膜向负极方向迁移,在负极上捕获1个电子被还原为Li并存贮在具有层状结构的石墨中。放电时在负极中锂会失去1个电子而成为锂离子Li+,Li+穿过隔膜向正极方向迁移并存贮在正极材料中[1-2,5]。由于在充放电时锂离子是在正负极之间来回迁移,因此称之为“锂离子电池”。
1.2 特点
锂离子电池发展迅速、应用广泛,这与锂离子电池固有的特点是密切相关的。现将锂离子电池与其他电池的性能比较如表1。
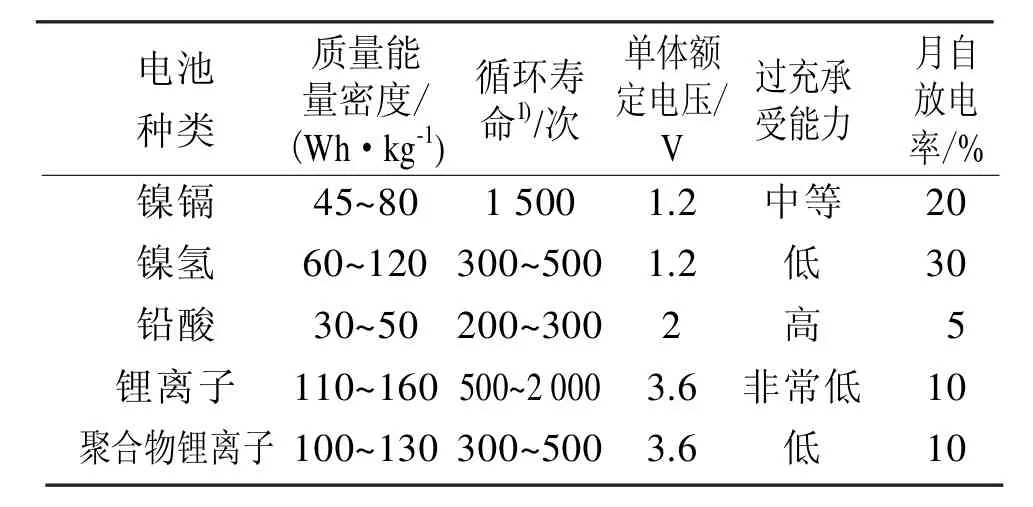
表1 锂离子电池与其他电池性能比较Tab 1 Performance comparison of lithium batteries and other batteries
由表1可知,锂离子电池与Ni/Cd、Ni/MH、铅酸电池相比,具有以下优点[1,3,6]:
1)工作电压高(3.6 V)。为Ni-Cd和Ni-MH电池(1.2 V)的 3倍;
2)能量密度高,开发潜力大。UR18650型的体积容量和质量容量分别可达300 W·h/cm3和125 W·h/kg。由于实际能量密度与理论值还有较大差距,因此锂离子电池的能量密度仍有可提高的空间;
3)循环寿命长,安全性好。由于锂离子电池使用的是嵌锂碳材料而不是金属锂,因此在锂离子电池充放电过程中,锂离子在正、负极中有相对固定的空间和位置,而不象一次锂电池那样沉积在金属锂负极表面,从而避免了树枝状枝晶的形成而造成的内部短路,使电池充放电反应的可逆性很好,保证了电池的长循环寿命和工作的安全性;
4)绿色环保,无记忆效应。锂离子电池的负极是嵌锂碳材料,没有毒性,正极是锂的过渡金属氧化物,毒性很小。同时电池被很好地密封,在使用过程中没有气体放出,是一种无毒无污染的电池体系。另外,它不像镍镉电池那样具有记忆效应;
5)自放电率低。锂离子电池在首次充放电过程中会在碳负极表面形成一层钝化膜,它允许离子通过但不允电子通过,因此可以较好地防止自放电,使电池的自放电率大大减小;
6)工作温度范围宽。锂离子电池采用的是有机电解液体系,它可以-30~60℃的温度内良好地工作;
7)输出功率大;
8)可快速充放电:1C充电时容量可达标称容量的80%以上。
当然,锂离子电池还有以下一些不足之处:
1)成本高,主要是正极材料、隔膜、电解液价格高;
2)必须有特殊的保护电路,以防过充电;
3)与普通电池的相容性差,因为一般要在用3节普通电池的情况下才能用锂离子电池代替。
2012年1月工信部发布新材料产业 “十二五”发展规划指出,2015年,新能源汽车累计产销量将超过50万辆,需要能量动力电池模块15 GWh/a、功率型3 GWh/a、电池隔膜108m2/a、六氟磷酸锂电解质盐1 000 t/a、正极材料10 kt/a、碳基负极材料4 000 t/a。规划确立实施“先进电池材料专项工程”:组织开发高效率、大容量、长寿命、安全性能高的磷酸盐系、镍钴锰三元系、锰酸盐系等锂离子电池正极材料,新增正极材料产能45 kt/a,推进石墨和钛酸盐类负极材料产业化,新增负极材料产能20 kt/a。可以预见先进电池材料的研发与应用异常活跃,将成为竞争热点,它对于电动汽车的商业化具有决定性的作用。而锂离子电池正极材料是锂离子电池的核心,正极材料的好坏决定着锂离子电池性能的优良与否,因此寻找具备优良性能的正极材料是锂离子电池发展的关键。
以下重点讨论锂离子电池正极材料的国内外现状及发展方向。
2 研究进展
2.1 现状
在整个锂电池产业链目前的产能比较中,由于进入壁垒较高,锂电池正极材料的产能是最小的,所以成为最关键的一个环节。能应用于锂离子电池中做正极材料的有很多种,目前研制成功并得到应用的多为过渡金属嵌锂化合物,大致可分为3种结构:1)六方层状结构,代表材料包括 LiCoO2、LiNiO2、Ni、Co、Mn 复合氧化物,三元材料 LiNi1-xyCoxMnyO(0≤x,y≤1,x+y≤1);2)尖晶石结构,代表材料为 LiMn2O4;3)橄榄石结构,代表材料为LiFePO4等,见图1。

目前商业化的几种锂离子电池正极材料性能优劣见表2。
2.1.1 钴酸锂
一直以来,LiCoO2在正极材料市场上占有主导地位,其制备基本上采用固相法,即Li2CO3和Co3O4机械混合后高温烧结,控制烧结条件,如温度、时间、保护气氛等,以得到所需颗粒的粒度、形貌和比表面等。
钴酸锂具有制备工艺简单、开路电压高、比能量大、循环寿命长、能快速充放电、电化学性能稳定等优点。同时钴酸锂存在着一些缺点,如实际容量与理论容量相差太大;正常充电结束后,LiCoO2正极材料中的Li还有剩余,埋下了使电池内部短路的安全隐患;Co的成本高、毒性大、环境污染较大等。同时LiCoO2为层状结构属于六方晶系,具有R3m空间群。
LiCoO2结构单元中,O2-为···ABCABC···面心立方最紧密堆积排列,Li+和Co3+交替占据立方紧密堆积的八面体间隙的3a和3b位。实际上由于Li+和Co3+与O2-离子层作用力的不同,导致O2-的分布偏离理想的最紧密堆积结构而呈现三方对称。所以在高电压LiCoO2的结构不稳定,循环性能差,且Co4+、O2-活性大,容易引发安全事故。LiCoO2理论比容量为274 mAh/g。当Li+脱出超过50%时,脱锂态的Li1-xCoO2(x<0.55)具有较高的氧化性,会导致解液分解和及流体腐蚀[7]。此外,Li+从CoO2层间脱出,会引起CoO2层间排斥力增大,六方晶体沿c轴膨胀,使CoO2发生三方晶系与单斜晶系的转变[8]。Li+从LiCoO2中可逆脱/嵌量最多为0.5个单元,因此,LiCoO2实际容量与理论容量相差太大只有130~150 mAh/g。
针对LiCoO2存在的一些问题,人们在LiCoO2的基础上对钴酸锂进行了改性如掺杂元素以提高LiCoO2的晶体稳定性,通过表面包覆提高高电压下高活性离子的稳定性,降低电池滥用条件下的热效应。
Idemoto等分别采用固相法和溶胶-凝胶法合成了LixNi0.8Co0.2O2[9]。研究发现,采用固相法合成的产物具有较好的电化学和循环性能,原因是用固相法制备的LixNi0.8Co0.2O2材料中Li含量低,阳离子混排程度小,Li+的扩散性好。进一步研究表明,Li含量与合成方法都会对材料结构中3a-6c和3b-6c间的结合力产生影响,进而影响到LixNi0.8Co0.2O2材料结构的稳定性和电化学性能。
Yang等以NH4HCO3为沉淀剂,采用新的共沉淀合成路线合成了LiNi0.8Co0.2O2正极材料[10]。与传统的共沉淀路线相比,前者合成的LiNi0.8Co0.2O2正极材料表现出良好的电化学和循环性能,初始放电比容量达到194.8 mAh/g,20次循环后,容量保持率达到91.9%。采用共沉淀法合成的材料具有良好而规整的层状结构,且合成的材料为亚微米尺寸颗粒,粒径分布较窄,有利于Li+的脱嵌。

表2 常用几种正极材料性能比较Tab 2 Performance comparison of several kinds of positive pole materials
2.1.2 尖晶石锰酸锂
与锂钴氧化物和锂镍氧化物相比,锂锰氧化物安全性好、耐过充性好、工作电压高,而且锰资源丰富、价格低廉、无毒,因此,Li-Mn-O系化合物是锂离子电池中最具发展前景的正极材料之一。
尖晶石型LiMn2O4属于立方晶系,Fd3m空间群。在LiMn2O4结构单元中,O2-里面心立方紧密堆积,Li位于密堆积的四面体间隙,构成LiO4框架,Mn位于八面体间隙,构成MnO6框架,整个结构由[LiO4]四面体和[MnO6]八面体构成。其结构的每个顶角为1个四面体和3个八面体共有,四面体的1个面和3个顶点与1个空的八面体构成Mn2O4网络框架。每个LiMn2O4晶胞包含8个LiMn2O4结构单元。根据晶体结构理论,在球的最密堆积中,四面体间隙数为球数的2倍,八面体间隙数与球数相同,所以一个LiMn2O4晶胞由32个O2-构成面心立方最密堆积,形成64个四面体间隙和32个八面体间隙[11]。8个锂离子占据四面体间隙的1/8(8a),16个锰离子占据八面体间隙的1/2(16d),且Mn3+和Mn4+各占一半,剩余的56个四面体间隙(8b和48f)和16个八面体间隙16C全空,这些空的间隙构成了Li+扩散的三维通道,因此该结构比层间化合物更有利于Li+离子脱嵌。
目前锰酸锂的主要制备方法有[12]:
1)固相烧结法:固相烧结法是最早用于制备尖晶石LiMn2O4的方法,该方法是将氢氧化锂或锂盐与锰的氧化物或锰盐按一定比例混合,研磨、烧结或多次研磨再烧结的方式制得粉体。其特点是合成时间较长,产物均匀性稍差,但该方法工艺简单,易于实现工业化,为国际上生产锰酸锂的厂家普遍采用[13]。
2)熔盐浸渍法:熔盐浸渍法是改进了的固相合成法,它采用低熔点的Li(OH)·H2O或 LiNO3作锂源,在高于锂盐熔点的温度烧结,使熔融的锂盐浸渍到锰化合物的表面,增加反应物之间的接触,提高反应效率。Yongyao Xia等采用该方法制备的材料首次放电比容量为135 mAh/g,循环性能也较为理想[14]。但是LiNO3在烧结过程中释放出NO2气体,对作业环境危害较大。而Li(OH)·H2O由于含有结晶水,原料的配比难以掌握。
3)微波合成法:介质材料放在微波场,介质材料中新形成的偶极子或原有的偶极子在高频交变电磁场中发生重排,产生类似摩擦的作用而产生大量的热。该方法可实现均匀受热、快速升温,大大缩短反应时间,制备的材料也具有优良的电化学性能,首次放电比容量可达120 mAh/g以上[15-16]。
4)溶胶-凝胶法:溶胶凝胶法中最为典型的是Pechini法,它基于金属离子与有机酸能形成螯合物而把锰离子和锂离子同时螯合在大分子上,再脂化进一步形成均相固态高聚物前驱体,然后烧结前驱体制得材料[17]。由于锂离子、锰离子和其他金属离子被同时螯合在一个大分子上,它们之间的距离非常小,可以克服氧化物形成过程中的远程扩散,因此,合成温度低。该方法制备的材料具有优良的电化学性能,首次放电比容量可达130 mAh/g[18]。
5)共沉淀法:共沉淀法则是将锂盐和锰盐混合溶解后,使锂和锰同时以沉淀的形式析出,将沉淀干燥制得均相前驱体,再将前驱体烧结即可得到材料。采用共沉淀法制备的材料颗粒细小,成分均一化程度高,但是反应过程不易控制[19]。由于锰的化合价复杂,嵌锂氧化物多种多样,不同的合成方法及反应条件对材料的结构和性能影响很大。
虽然LiMn2O4是理想的4 V级正极材料,但是其存储性能较差、容量衰减较快,尤其是高温(>55℃)循环性能较差[20]。经研究得出能引起LiMn2O4容量衰减的主要原因有:
1)放电末期Mn3+发生歧化反应:生成的Mn2+溶于电解液中,溶解的Mn2+会扩散到负极碳材料表面,导致不可逆容量损失:

2)Jahn-Teller效应,锂离子在嵌入、脱嵌时LiMn2O4发生结构变化,[MnO6]歧变,使结构发生变化并伴有更大的压缩和拉伸应变差,导致材料性能衰减。
3)充电生成的Mn4+在电解液中不稳定。
4)在使用LiPF6电解质的电解液时,高温下(55~65℃)电解液中少量的H2O与锂盐生成HF,HF与LiMn2O4发生反应,导致Mn2+溶解:

并且LiMn2O4还具有催化电解液分解的作用,加剧了材料性能的衰降。在提高尖晶石LiMn2O4循环性能方面,主要有体相掺杂和表面包覆2种方法。掺杂不仅可以提高晶格的无序化程度,增强尖晶石结构的稳定性,而且当掺杂离子的价态小于或等于+3时会降低Mn3+离子的含量,使Mn4+/Mn3+增大,锰的平均化合价升高,抑制Jahn-Teller效应。
表面包覆是在LiMn2O4颗粒表面覆盖一层其他材料,减少电解液与LiMn2O4的接触面积,减少LiMn2O4的溶解。表面包覆可以保持材料的原有结构和粒度,在掺入量较低的前提下对颗粒表面进行高浓度的掺杂,达到高含量掺杂的效果。Co3+的离予半径(0.068 nm)与 Mn3+的离子半径(0.064 5 nm)接近,Co掺杂后Mn的平均化合价升高,在一定程度上减少Jahn-Teller的发生。
Co3+的电子构型(t62geog)具有比 Mn(t32ge1g)更强的配位场稳定能,更强的M—O键能(Mn—O键能946 kJ/mol,Co—O 键能为 1 067 kJ/mo1),在充放电过程中,晶体体积变化小(<5%),尖晶石结构更加稳定[21]。
Shen等报道,Co的掺杂使尖晶石颗粒增大,比表面积减少一半,大大抑制了容量的衰减,改善了材料的循环性能,掺Co后增加了交换电流密度,有助于电荷转移[22]。电池正极材料的电子授受大部分由阴离子承担,阴离子对正极材料性能的影响至关重要[23]。据此,阴离子掺杂也是可能改善尖晶石LiMn2O4性能的一条途经。氟的电负性比氧大,吸引电子能力强,能稳定材料的结构。Amatucci G G等认为F-取代O2-使Mn的平均化合价降低,使部分Mn4+还原为Mn3+,可以提高材料的放电比容量[24]。
最新研究表明,单一的F掺杂可以提高材料的放电比容量,但是F取代O后,锰的平均化合价降低,导致Jahn-Teller畸变,使材料循环性能变差[25]。为了补偿该影响,使锰的平均化合价在+3.5以上。Amatucci等同时掺杂了Al、F,提高了锰的价态,不仅提高了电极材料在高温稳定性,而且还有效抑制了锰在电解液中的溶解[24]。
体相掺杂可以改善LiMn2O4的循环性能,但是掺杂含量稍大就可能引起材料的初始比容量明显减少,要抑制锰的溶解和电解液在电极上的分解,提高LiMn2O4在较高温度下的电化学性能,表面包覆是另外一种行之有效的方法。基于CeO2具有较强的催化能力且与氧化物有良好的电接触。Hyung-Wook Ha等研究了CeO2包覆的LiMn2O4,结果显示,质量分数2%的CeO2包覆的尖晶石LiMn2O4具有良好的高温稳定性,60℃、0.5C倍率首次放电比容量为 117 mAh/g,循环40周期后容量保持率为82%,而未经包覆的材料的容量保持率为59%[26]。
由于TiO2结构中有存储小离子(如H+、Li+)的通道,Lihong Yu等采用溶胶凝胶法制备了TiO2包覆的尖晶石LiMn2O4,TiO2包覆后材料的首次放电比容量有所提高[27]。
2.1.3 磷酸铁锂
具有橄榄石结构的LiFePO4。在自然界中以磷铁锂矿的形式存在,属于正交晶系,空间群为Pmnb。每个晶胞中有4个磷酸铁锂单元,其晶胞参数为:a=0.600 8 nm,b=1.032 4 nm,c=0.469 4 nm。
图2为LiFePO4结构及Li+迁移示意。
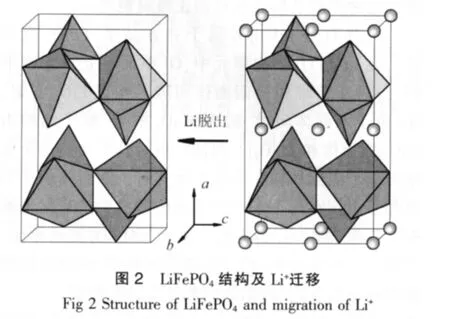
在LiFePO4中,氧原子近似于六方紧密堆积,磷原子占据PO4四面体4c位,铁原子和锂原子分别占据八面体4c和4a位。从b轴方向的视角出发,可以看到FeO6八面体在6c平面上以一定的角度连接起来。而LiO6八面体则沿b轴方向相互共棱,形成链状。1个FeO6八面体分别与1个PO4四面体和2个LiO6八面体共棱。同时,1个PO4四面体还与2个LiO6八面体共棱。Li在4a位形成共棱的连续直线链,并平行于c轴,从而Li+具有二维可移动性,使之在充放电过程中可以脱出和嵌入。
强的P—O加共价键形成离域的三维立体化学键,使磷酸铁锂具有很强的热力学和动力学稳定性。LiFePO4在充放电过程中,Li+的可逆嵌脱,对应于Fe3+/Fe2+的互相转换,电压平台在 3.5 V(vs.Li+/Li),且平台较长。由于P—O键键强非常大,所以PO4四面体很稳定,在充放电过程中起到结构支撑作用,因此LiFePO4有很好的抗高温和抗过充电性能,同时由于LiFePO4和完全脱锂状态下的FePO4的结构很相近,所以LiFePO4的循环性能也很好。
磷酸铁锂理论比容量较高,为170 mAh/g,相对金属锂的电压为3.4 V,在小电流充放电时有着极为平稳的充放电平台。这主要是在LiFePO4晶体中的2个Fe原子和1个P原子共用1个O原子,Fe—O—P的诱导效应削弱了Fe—O键的强度,而聚阴离子团PO43-使LiFePO4结构稳定,并降低了Fe3+/Fe2+氧化还原电对的费米能级,从而增加电极电位。充放电过程是在LiFePO4和FePO4两相之间进行,两相互变过程中Fe—O和P—O原子之间距离变化不大,在充放电过程中该材料体积变化较小,这种变化正好与碳负极在充放电过程中所发生的体积变化相抵消,总体上并没有影响其电化学性能的体积效应产生。其次,LiFePO4与FePO4在结构上极为相似,在锂离子的脱嵌过程中,晶体结构不需要发生重排,这也是LiFePO4具有优异循环性能的一个主要原因。
在磷酸铁锂中,锂与氧之间是共价键结构,这一特性决定了磷酸铁锂在高温下难以释放出氧,大大提高了它的热稳定性。尽管LiFePO4有着众多优异性能,但位于LiO6八面体和FeO6八面体之间的PO4四面体限制了LiFePO4的体积变化,影响锂离子在充放电过程中的嵌入和脱出,使得LiFePO4的离子扩散率较低,为 10-14~10-11cm2/s[28]。 其次,在 LiFePO4结构中没有连续的FeO6共棱八面体网络,不能够形成电子导体,电子传导只能通过Fe—O—Fe进行,使LiFePO4电子电导率较低,为10-9S/cm[29]。电子迁移率低反过来又会以极化的形式影响锂离子的扩散。
目前合成LiFePO4的主要方法有[30]:
1)高温固相反应法。高温固相反应法可分为一步加热法、二步加热法和三步加热法。其中二步加热法最常见,即将原料充分研磨,在惰性气氛中预热处理,之后再研磨,于惰性气氛中烧结为最终产品。一步加热法省去了预热处理工序,而三步加热法是在预热处理工序之后增加一步烧结工序(即采用2段温度烧结)。固相法是目前制备LiFePO4最常用、最成熟的方法。研究结果表明,采用均相前驱体,以中等温度(500~600℃)焙烧,得到产物的放电比容量在室温下可达160 mAh/g。焙烧温度大于800℃时,所得产物粒径较大、比表面积较小、导电性能差。
2)水热合成法。水热法是以可溶性亚铁盐、锂盐和磷酸为原料,在水热条件下直接合成LiFePO4。由于氧气在水热体系中溶解度很小,水热体系为LiFePO4的合成提供了优良的惰性环境,因此水热合成不再需要惰性气体保护。水热法可以直接合成LiFePO4,产物晶型和粒径易于控制,但水热法需要耐高温高压设备,工业化生产难度较大。S Yang等以可溶性2价铁盐、LiOH和H3PO4为原料,在120℃下采用水热法在短时间(5 h)内合成了LiFePO4,平均粒径约为3 μm[31]。这种材料以0.14 mA/cm2的电流密度充放电,比容量为100 mAh/g。在该合成研究中,使用氢氧化锂(LiOH)作为沉淀剂,需要多消耗200%的LiOH,从而增加了原料成本,选用其他廉价沉淀剂,如氨水、碳铵、尿素等,也是将来工艺改进的方向之一。
3)溶胶-凝胶法是以3价铁的醋酸盐或硝酸盐为前驱体,混合化学计量的LiOH后加入柠檬酸,然后加入到H3PO4中,采用氨水调节pH,加热至60℃得到凝胶。在300~400℃加热12 h使凝胶分解,最后在700~800℃烧结得到LiFePO4。该法优点是前驱体溶液化学均匀性好、凝胶热处理温度低、粉体粒径小而且分布窄、粉体烧结性能好、反应过程易于控制、设备简单。缺点是干燥收缩大、工业化生产难度较大、合成周期较长。
P P Prosini等用过氧化氢氧化六水硫酸亚铁铵与磷酸二氢铵的混合物,在水中合成亚稳态的无定形FePO4,再用LiOH等按照化学计量以化学方法锂化 FePO4生成 LiFePO4[32]。
4)氧化还原法。氧化还原法是将可溶性Fe(Ⅱ)氧化成Fe(Ⅲ)使之形成FePO4沉淀,然后用化学方法把FePO4还原成LiFePO4。该方法制得的磷酸铁锂晶粒为纳米级且粒径分布均匀,但工艺复杂,不能大量生产,只适用于实验室研究。P P Prosini等以NH4H2PO4和Fe(NH4)2(SO4)2·6H2O为原料,在溶液中用 H2O2把 Fe(Ⅱ)氧化成 Fe(Ⅲ),使之形成 FePO4沉淀,然后将过滤所得沉淀物浸泡在1 mol/L的LiI溶液中(乙腈作溶剂),持续搅拌使之反应24 h,过滤,在550℃下焙烧1 h,即得产物。制得的产物以0.1C和3C倍率充放电,其放电比容量分别为164 mAh/g和140 mAh/g[33]。
目前改善LiFePO4的方法主要有:1)合成粒径小且均匀的颗粒;2)包覆导电剂对颗粒表面进行改性;3)掺杂高价金属离子等。目前的表面包覆材料主要集中于碳和金属粉末。由于碳具有良好的导电性能和成本低廉的优势,很多学者都采用碳包覆的方法来改善LiFePO4的性能。
Z H Chen等合成的含碳质量分数为3.5%的LiFePO4在0.1C下放电,常温下容量可达160 mAh/g,接近其理论容量[34]。不同的碳源对材料的改性效果不同。蒋永等以苯为碳源,采用气相沉积法得到LiFePO4样品,在0.1C下首次放电比容量达到151.6 mAh/g,1C下首次放电比容量达到125.8 mAh/g[35]。
2.2 纳米技术应用
目前应用的大部分锂离子电池正极材料或多或少的在某些方面存在着缺陷,寻求新的技术突破,找到足够高的嵌锂容量和很好的锂脱嵌可逆性,以保证电池的高电压、大容量和长循环寿命的要求的正极材料。纳米材料具有特殊的物理、化学性能,在电子材料和催化剂等方面已经得到成功应用[36]。随着纳米材料的应用越来越广,电池研究者们开始将纳米材料应用到该领域。纳米正极材料能够很大程度提高嵌入、脱出速率,提高比功率,被认为是最有希望突破这些限制从而达到多元化目标,有很好发展潜力的材料。不同制备方法合成的纳米正极材料,其电化学性能有一定差别,因此对纳米合成方法的研究显得更有意义。
10多年来,电池工作者致力于锂离子电池纳米电极材料的研究,并且取得了一定的进展,如:Chen等研究了粒径在20~100 nm的LiCoO2,该材料具有良好的电化学性能,特别是其高倍率充放电性能优越,在50C的放电倍率下其放电比容量可达到100 mAh/g[37];Pan等研究了纳米结构的Li3V2(PO4)3/C材料(粒径<50 nm),在电压 3~4.3 V内循环,速率为1C时可逆的放电比容量达到122 mAh/g,在32C的速率下放电比容量维持在83 mAh/g,证明纳米结构的Li3V2(PO4)3和多孔隙碳组成的正极材料应用于高功率锂离子电池中有很大潜力[38]。
具有一维隧道结构的嵌锂化合物LiFePO4虽然具有良好的充放电平台和安全性能,但导电率低和离子扩散性能差等严重影响了其倍率性能的提高[39]。研究发现,前者通过添加导电剂已基本解决达到实验的水平,减小粒子的尺寸到纳米级可以增强倍率性达到实际应用水平[40-41]。
Wang等合成了以尺寸为20~40 nm的LiFePO4为核,厚度为1~2 nm的半石墨碳为壳的核壳结构,表现出了高倍率性和良好的循环性,在60C时放电比容量达到 90 mAh/g,0.6C下比容量为168 mAh/g,循环1 100周后放电比容量损失小于5%[42]。
2.3 纳米正极材料的合成方法
2.3.1 固相法
固相法因简单方便、容易操作,是大规模生产的最佳方法,成为无机材料合成中最常用的方法之一。夏熙等用低热固相反应法合成纳米LiCoO2,发现了混配效应:以一定比例纳米LiCoO2与常规LiCoO2进行混配,制成的电池的充电容量可达132 mAh/g,放电容量为 125 mAh/g,放电平台在3.9 V,由于纳米颗粒增大了比表面积,令Li+更易嵌入和脱出,削弱了极化现象,循环性能比常规LiCoO2明显提高,显示出较好的性能[36]。
目前很多研究者在传统固相法结合其他方法方向进行了研究。王志高等在材料合成过程中采用超声波分散,在数分钟内合成了粒径约为500 nm的LiFePO4/C复合材料,微波烧结8 min的材料0.1C下的首次放电比容量可超过140 mAh/g[43]。碳的加入,可增强导电性,改善循环性能,但团聚较明显;快速合成时内部反应可能不充分,晶格结构不规整、不稳定,大电流循环时易出现结构崩塌,高倍率性能较差。
但是固相法的不足是普遍合成周期长、能耗较大、颗粒不均匀。固相法中原料的化学稳定性、热稳定性、熔点等性质对于合成纳米材料能否成功至关重要,因此原料的选择必须慎重[42]。
2.3.2 溶胶-凝胶法
溶胶-凝胶法是制备纳米材料的一种常用方法,广泛应用于金属氧化物纳米粒子的制备。该方法的优点在于:原料可达到原子水平均匀混合、合成温度低,产品的化学性能好、粒径小且分布窄、比表面积大、晶体结晶程度高。前驱物用金属醇盐或非醇盐均可。其实质是前驱物在一定条件下水解成溶胶,再制成凝胶,经干燥和热处理后制得所需纳米粒子。
伍昌维等采用溶胶-凝胶法成功制备纳米级电池正极材料LiMn2-xAgxO4,且在850℃煅烧制备的正极材料LiMn22-xAgxO4的颗粒呈规则的六方粒状,平均粒径约为100 nm。由扫描电子显微镜(SEM)、透射电镜(TEM)和原子力显微镜(AFM)、能谱(EDS)分析均可得出材料的粒度分布范围均在纳米级[44]。
Lee等以脂肪酸为螯合剂,通过溶胶-凝胶法合成了纳米LiFePO4晶体颗粒,未进行任何的表面处理及包覆[45]。首次放电比容量高于150 mAh/g,循环70次后容量几乎没有衰减,且在30C的高倍率下仍具有很好的循环稳定性。目前制得了多形态纳米级的粉状、纤维状、多孔LiFePO4正极材料以及薄膜正极材料,其电化学性能明显优于固相法,这可能溶胶凝胶法制备得到的样品成分更为均一、纯净,同时与在热处理过程中取向生长有关[42]。
2.3.3 冷冻干燥法
由于纳米粒子的表面效应,电极材料前驱体在干燥过程中容易团聚,因此采用适宜的干燥技术是必要的,冷冻干燥法是一种合成均匀分散的纳米级LiFePO4/C复合材料的有效方法。
V Palomares等用柠檬酸作为络合剂和碳源,经过冷冻干燥的溶液在低温度的热处理后形成均匀的碳包覆LiFePO4样品,粒径小于100 nm,循环伏安测试显示可逆性很好,在0.025C放电比容量达到164 mAh/g,1C时为146 mAh/g,且循环50周后容量保持率为97%,表明冷冻干燥法是制备锂离子电池纳米正极材料的一种积极有效的方法[46]。由于有工业装置造价高、操作周期长、能耗大而且设备效率低等缺点,实现工业化有一定的难度。
2.3.4 微乳液法
微乳液法是利用金属盐和一定的沉淀剂在水、油、表面活性剂形成的微乳液体系中反应,在其水核(称为微反应器)微区内控制胶粒成核生长,热处理后得到纳米微粒。此法得到的纳米颗粒具有粒径分布窄、呈球形或椭球形以及分散性好等优点。
Kim等用乳化剂Span80、煤油和石蜡油的混合物为乳化有机相,在不同温度下煅烧,得到粒径小于100 nm的LiNiO2颗粒[47]。在750℃煅烧,产物的电化学性能最好,首次循环的放电比容量为161 mAh/g,20次循环后的容量保持率达91%。
产物晶核生长局限在乳液的水核内部,可通过水核来控制产物的大小和形状,使形成的材料粒度可控,容易得到纳米级细小颗粒,材料的结构稳定且纯度高,电化学性能优良。该方法的制备过程较复杂,成本较高。
2.3.5 其他方法
郭玉国等提出了“分级三维(3D)混合导电网络”电极材料设计,为开发高能量、高功率动力型锂离子电池电极材料提供了新途径[48]。方法是利用纳米孔道和纳米电子导电网络分别实现纳米尺度上Li+和电子的快速输运,从而构筑出纳米级的3D离子/电子混合导电网络;再以该纳米网络作为结构基元,并与导电添加剂复合形成微米级的3D混合导电网络,从而实现锂离子和电子的高速输运与存储。并且成功把LiFePO4纳米颗粒分散在多孔碳3D导电网络中,开发出“3D混合导电网络”结构高倍率正极材料[49-50]。 参见图3。PO4/C样品。采用差热-热损失、X射线衍射、扫描电镜、比表面测试、电化学性能测试等分析测试方法对纳米FePO4·xH2O、FePO4前驱体及不同煅烧温度下制得的纳米LiFePO4/C样品进行表征[51]。结果表明,700℃烧结10 h合成LiFePO4/C样品的粒径在40~100 nm左右,比表面积为79.8 m2/g;700℃煅烧合成样品在电压 2.5~4.2 V, 倍率为 0.1C、1C、5C、10C、15C时的放电比容量分别达到156.5、134.9、105.8、90.3、80.9 mAh/g,具有较好的倍率性能,样品还表现出较好的容量保持率。
通过上述锂离子电池纳米正极材料各种合成方法,可以看出,经过研究者们不断努力和大胆尝试,一些合成具有新特性或者复合物特性的纳米材料的新型方法层出不穷,合成的材料表现出良好的电化学性能,成为制备纳米级锂离子电池正极材料的一类积极有效的方法。有些方法基于它们的原材料、设备等条件较昂贵减缓了实用化的进程;喷雾干燥法、溶胶-凝胶法和水热法等是实验室合成纳米正极材料的常用方法,能通过实验参数来控制材料的结构,易得到纳米级结构稳定、电化学性能好的纯相材料。
但应用到生产中出现了一些问题,如喷雾干燥法固气比很低、设备体积庞大、体积传热系数小、热效率较低、凝胶的生成周期长、干燥后体积收缩大;水热法对实验参数的控制要求严格,这些都影响了大规模生产;但是这些方法的研究提供了很多解决问题的思路,最大可能地发挥各种方法的优点尽量弥补不足。
3 总结

武玉玲等以Fe为铁源,采用控制结晶技术合成了纳米 FePO4·xH2O, 将 FePO4·xH4O 于 500℃热处理4 h后得到纳米FePO4前驱体,然后通过碳热还原在不同温度下煅烧合成橄榄石结构的纳米LiFe-
目前国内应用的正极材料主要有钴酸锂、锰酸锂和磷酸铁锂等,并且针对这些材料的一些缺陷进行了大量的改性工作,一些改性后的工艺如掺杂金属离子,表面包覆等也得到了应用,但是总体上还有很大的进步空间。而纳米技术应用于电池正极材料给锂电池正极材料的发展提供了很好的突破方向。通过电池工作者10多年来的研究发现,锂离子纳米正极材料有如下的优点:尺寸小,Li+嵌脱路径短,能更好地释放嵌脱锂的应力,加速Li+扩散,提高快速充放电能力;表面张力比普通正极材料大,嵌锂过程中,溶剂分子难以进入材料的晶格,因此可阻止溶剂分子的共嵌,延长电池的循环寿;比表面积较大,与电解液的接触面积大,能提供更多的Li+嵌脱位置;表面的高孔隙率也使嵌锂空位增多,具有比普通正极材料更高的容量;超塑性和蠕变性使得其对体积变化具有较强的承受能力并降低聚合物电解质的玻璃化转变温度。
纳米正极材料的这些优点,使其具有现实意义和广阔的发展前景。如根据现有正极材料出现的问题,利用优化设计改进传统的合成方法,通过掺杂、包覆、加入辅助剂和表面修饰改性等方法减低成本,利用纳米材料的优点和微米材料优良的稳定性和容易制备的优点合成纳微分层结构的材料(如纳米棒自组装成的微米球)解决纳米材料的低热力学稳定性、团聚及与电解液发生副反应等问题。可以尝试着探索新的方法合成纳米级颗粒,并将最优的方法应用于新材料和经典电极材料的制备,从而充分发挥纳米级材料的尺寸效应和表面效应,改善电极材料的电化学活性,有助于推进纳米正极材料的工业化进程。
[1]郭炳琨,徐徽,王先友,等.锂离子电池[M].长沙:中南大学出版社,2002.
[2]吴宇平,万春荣,姜长印,等.锂离子二次电池[M].北京:化学工业出版社,2002.
[3]吴宇平,戴晓兵,马军旗,等.锂离子电池[M].北京:化学工业出版社,2004.
[4]陈立泉.电动车锂离子电池的材料问题[J].中国工程科学,2002,4(11):32-36.
[5]郑子山,张中太,唐子龙,等.锂离子二次电池最新进展及评述[J].化学世界,2004(5):270-273.
[6]姚晓林.锂离子电池关键材料的电化学性能及热稳定性研究[D].合肥:中国科学技术大学,2005.
[7]M G S R Thomas,P G Bruce,J B Goodenough.AC impedance of the Li(1-x)CoO2Electrode[J].Solid State Ionics,1926,18(2):794-798.
[8]D Carlier,A Van der,Van C Delmas,et al.First-principles investigation of phase stability in the O2-LiCoO2system[J].Chemistry of Materials,2003,15(13):2651-2660.
[9]Idemoto Y,Takanashi Y,Kitamura N.Dependence of property,crystal structure and electrode characteristics on Li content for LixNi0.8Co0.2O2as a cathode active material for Li secondary battery[J].J Power Sources,2009,189:269-278.
[10]Yang Z X,Wang B,Yang W S,et al.A novel method for the preparation of submicron-sized LiNi0.8Co0.2O2cathode material[J].Electrochim Acta,2007,52(28):8069-8074.
[11]张克从.近代晶体学基础[M].北京:科学出版社,1987:226-228.
[12]马尚德.锂离子电池正极材料尖晶石锰酸锂的制备与改性研究[D].长沙:中南大学,2007.
[13]谭柱中,梅光贵,李维健,等.锰冶金学[J].长沙:中南大学出版社,2004:638-649.
[14]Yongjao Xia,Yasufumi Hideshima,Naoki Kumade,et al.Studies on Li-Mn-O spinel system(obtained from meltimpregnation method)as a cathode for 4 V lithium batteries:Part V.Enhancement of the elevated temperature performance of Li/LiMn2O4cells[J].Journal of Power Sources,1998,74(1):24-28.
[15]彭虎,李俊.微波高温加热技术进展[J].材料导报,2005,19(10):100-103.
[16]Ben-Lin He,Wen-Jia Zhou,Shu-Juan Bao,et al.Preparation and electrochemical properties of LiMn2O4by the microwave-assisted rheological phase method[J].Electrochimica Acta,2007,52(9):3286-3293.
[17]Liu W,Farriagton G C.Synthesis and electro-chemical studies of spinel phase LiMn2O4cathode materials prepared by the pechini process[J].Journal of the Electrochemical Society,1996,143(3):879-884.
[18]何向明,王莉,张国昀.溶胶凝胶法合成锂离子电池正极材料LiMn2O4[J].电化学,2006,12(1):104-106.
[19]X Qiu,X Sun,W Shen,et al.Spinel Lil+xMn2O4synthesized by co-precipitation as cathodes for lithium ion batteries[J].Solid State Ionics,1997,93(3/4):335-339.
[20]Yongyao Xia,Masaki Yoshio.Studies on Li-Mn-O spinel system(obtained from melt-impregnation method)as a cathode for 4 V lithium batteries Part IV.High and low temperature performance of LiMn2O4[J].Journal of Power Sources,1997,66(1):129-133.
[21]N Amdouni,F Gendron,A Mauger,et al.LiMn2-yCoyO4(0≤y≤1)intercalation Compounds synthesized from wetchemical route[J].Materials Science and Engineering:B,2006,129(1/3):64-75.
[22]C H Shen,R S Liu,R Gundakaram,et al.Effect of Co doping in LiMn2O4[J],Journal of Power Sources,2001,102(1/2):21-28.
[23]宁连才,吴金平,周成刚,等.第一原理计算过渡金属掺杂尖晶石型LiMn2O4的电子结构[J].地球科学-中国地质大学学报,2006,31(3):317-320.
[24]Amatucci G G,Pereira N,Zheng T,et al.Failure mechanism and improvement of the elevated temperature cycling of LiMn2O4compounds through the use of the LiAlxMn2-xO4-zFzsolid solution[J].Journal of the Electrochemical Society,2001,148(2):A171-A182.
[25]J T Son,H G Kim.New investigation of fluorine-substituted spinel LiMn2O4-xFxby using sol-gel process[J].Journal of Power Sources,2005,147(1/2):220-226.
[26]Hyung-Wook Ha,Nan Ji Yun,Keon Kim.Improvement of electrochemical stability of LiMn2O4by CeO2coating for lithium-ion batteries[J].Electrochimica Acta,2007,52(9):3236-3241.
[27]Lihong Yu,Xingping Qiu,Jingyu Xi,et al.Enhanced high-potential and elevated-temperature cycling stability of LiMn2O4cathode by TiO2modification for Li-ion battery[J].Electrochimica Acta,2006,51(28):6406-6411.
[28]Prosini P P,Lisi M,zane D,et al.Determination of the chemical diffusion coefficient of lithium in LiFePO4[J].Solid state Ionics,2002,148(1/2):45-51.
[29]Chung S Y,Bloking J T,chiangm Y M.Electronically conductive Phosphor-olivines as lithium storage electrodes[J].Naature Materials,2002,l(2):123-128.
[30]冯国彪,邓宏.锂离子电池正极材料磷酸铁锂研究进展[J].无机盐工业,2011,3(43):14-17.
[31]Yang S F,Zavalij P Y,Whittingham M S.Hydrothermal synthesis of lithium iron phosphate[J].Electrochem Commun,2001,3(9):505-508.
[32]Prosini P P,Lisi M,Scaccia S,et al.Synthesis and characterization of amorphous hydrated FePO4and its electrode performance in lithium batteries[J].J Electronchem Soc,2002,149(3):A297-A301.
[33]Prosini P P,Zane D,Pasquali M.Improved electrochemical performance of a LiFePO4-based composite cathode[J].Electrochem Acta,200l,46:3517-3523.
[34]chen Z H,Dahn J R.Reducing carbon in LiFePO4/C composite electrodes to maximize specific energy,volumetric energy and tapdensity[J].J Electrochem Soc,2002,149(9):A1184-A1189.
[35]蒋永,赵兵,万小娟,等.气相沉积碳包覆磷酸铁锂的制备及性能[J].硅酸盐学报,2008,36(9):1295-1299.
[36]夏熙.纳米微粒作为电池活性材料的前景[J].电池,1998,28(6):251-254.
[37]Chen H L,Qiu X P,Zhu W T,et al.Synthesis and high rate proper-ties of nanoparticles lithium cobalt oxides as the cathode material for lithium-ion battery[J].Electrochem Commun,2002,4(6):488-491.
[38]Pan Anquan,Liu J,Zhang J G,et al.Nano-structured Li3V2(PO4)3/carbon composite for high-rate lithium-ion batteries[J].Electrochem Commun,2010,12(12):1674-1677.
[39]于锋,张敬杰,王昌胤,等.锂离子电池正极材料的晶体结构及电化学性能[J].化学进展,2010,22(1):9-18.
[40]李军,黄慧民,夏信德,等.锂离子电池纳米正极材料的研究进展[J].化工新型材料,2007,35(3):21-28.
[41]Peter G B,Bruno S,Jean-Marie T.Nanomaterials for rechargeable Lithium Batteries[J].Angew Chem Int Ed,2008,47:2930-2946.
[42]Wang Y G,Wang Y R,Hosono E,et al.The design of a LiFePO4/Carbon nano composite with a core-shell structure and its synthesis by an in situ polymerization restriction method[J].Angew Chem Int Ed,2008,47:7461-7465.
[43]王志高,李建玲,王新东.LiFePO4/C的微波法制备和性能[J].电池,2006,36(2):142-143.
[44]伍昌维,张朝平,龚宁.纳米级锂离子电池正极材料LiMn2-xAgxO4的制备[J].贵州化工,2009,34(5):1-4.
[45]Lee S B,Cho S H,Cho S J,et al.Synthesis of LiFePO4material with improved cycling performance under harsh conditions[J].Elec-trochem Commun,2008,10(9):1219-1221.
[46]Palomares V,Aintzane G,Izaskun G M.New freeze-drying method for LiFePO4synthesis[J].J Power Sources,2007,171:879-885.
[47]Kim B H,Kim J H,Kwon I H,et al.Electrochemical properties of LiNiO2cathode material synthesized by the emulsion method[J].Ceram Int,2007,33(5):837-841.
[48]郭玉国,王忠丽,吴兴隆,等.锂离子电池纳微结构电极材料系列研究[J].电化学,2010,16(2):119-124.
[49]Wu X L,Jiang L Y,Cao F F.LiFePO4Nan oparticles embedded in nan oporous carbon matrix:superior cathode material for electrochemical energy-storage devices[J].Adv Mater,2009,21:2710-2714.
[50]Liu C,Li F,Ma L P,et al.Advanced materials for energy storage[J].Adv Mater,2010,22:E28-E62.
[51]武玉玲,蒲薇华,任建国,等.纳米FePO4的合成及其正极材料LiFePO4/C的电化学性能研究[J].无机材料学报,2012,4(27):422-426.
Progress of Lithium ion Battery Positive Pole Material
Ming Bo,Han Hongyu
(Zhejiang Kaisn Fluorochemical CO.,LTD,Quzhou,Zhejiang 324004)
This paper introduced the development phase,work principle and characteristics of lithium ion battery,described the characteristics,synthesis method,advantages and disadvantages of the commercialized positive pole materials such as lithium cobalt oxide,lithium manganate and lithium iron phosphate,application and synthesis method of nanotechnology lithium ion battery positive pole material.The cost was reduced by doping,cladding;adding auxiliary agent and surface modification methods based on the problem existing in the pole material now available,nano differential layer structure materials were prepared using the advantages of nano material and excellent stability and easy preparation advantages of micron material which solved the problems of low thermodynamic stability in nano materials,reunion and side effects of the reaction with electrolyte;New methods to synthesize nanoscale particle can try to be explored,and the optimal method is applied in preparation of new materials and classical electrode materials,so as to give full play to nanoscale material size effect and surface effect,improve electrochemical activity of electrode material,promote the industrialization process of nano positive pole materials.
lithium ion battery;battery positive pole materials;lithium cobalt oxide;lithium manganate;lithium iron phosphate;nanomaterials;progress
TQ131.1+1
ADOI10.3969/j.issn.1006-6829.2012.04.009
2012-05-16
