原子吸收法简介(Ⅱ)(续完)
2012-12-06丘星初储荣邦
丘星初, 丘 山, 丘 圣, 储荣邦
(1.广州安纳环境分析测试有限公司,广东 番禺 511400;2.南京724研究所,江苏 南京210013)
原子吸收法简介(Ⅱ)(续完)
丘星初1, 丘 山1, 丘 圣1, 储荣邦2
(1.广州安纳环境分析测试有限公司,广东 番禺 511400;2.南京724研究所,江苏 南京210013)
叙述了原子吸收光谱分析法在电镀工业园区分析中的应用。简要介绍了原子吸收光谱分析的基本原理和原子吸收分光光度计的构造及其功能;着重介绍了原子吸收法中仪器最佳操作条件的选择,包括吸收波长、灯电流、预热时间、狭缝宽度、火焰性质、观测高度、燃烧器角度及试液提升量等,以便分析工作者根据仪器型号的不同进行优选。最后还介绍了仪器的日常使用与维护,及一般故障的排除方法。可供电镀工业园区实验室化验员参考。
原子吸收光谱分析法;电镀分析应用;仪器使用与维护
4 原子吸收法中的干扰及消除方法
原子吸收法中遇到的干扰有四类:化学干扰、电离干扰、光谱干扰和背景干扰。对各类干扰产生的原因及其消除方法分叙如下:
4.1 化学干扰及其消除
这类干扰是由于待测元素在空气-乙炔火焰中与干扰物生成难离解的化合物或高温难溶晶体所致。PO43-对 Ca2+的干扰,Al3+对 Mg2+的干扰及Si(Ⅳ)、Al3+、PO43-对Sr2+、Ba2+的干扰都是典型的化学干扰。其次,当Ca2+、Mg2+达到一定浓度,则会对 Cu2+、Zn2+、Pb2+及 Cd2+产生干扰。当 Cu2+、Ni2+、Mg2+的质量浓度达100mg/L时,会对不同质量浓度的Fe2+产生干扰。
消除化学干扰的方法有1)加入释放剂。即在试样中加入一种试剂,使其与干扰元素形成更稳定、更难离解的化合物,从而使待测元素从干扰元素形成的化合物中释放出来。如Al3+对Mg2+的干扰可加入La(NO3)3或SrCl2来消除。后者也常在消除 Si(Ⅳ)、PO43-等对 Ca2+、Sr2+、Ba2+、Mn2+及Fe2+的干扰中用作保护剂。一般加入量是1%La(NO3)3或LaCl3。
2)加入保护剂。通常使用配位剂作保护剂,使之与待测元素生成稳定的配位化合物而不再与干扰元素生成难离的化合物。常用的保护剂有EDTA和8-羟基喹啉,前者可消除Al3+和PO43-对Ca2+和Mg2+的干扰;后者可消除Fe2+对Cu2+、Ni2+和Mg2+等元素的干扰。
3)加入助熔剂。常用的是NH4Cl,因它熔点低,称之为助熔剂。它能提高某些元素测定的灵敏度,同时还可抑制Al3+、Si(Ⅳ)及PO43-对 Fe2+、Mn2+、Ca2+和Mg2+等的干扰。若在测定Cr(Ⅲ)或Cr(Ⅵ)时加入1%NH4Cl,不仅提高了Cr(Ⅲ)或Cr(Ⅵ)的灵敏度,还能有效地抑制Fe2+、PO43-等的干扰。
4)加入缓冲剂。在测定 20mg/L Ca2+时,H2SO4质量浓度大于0.01mg/L,H3PO4质量浓度大于0.05mg/L时干扰成定值。如果使标准系列和试液中都含有过量的这类干扰物质,虽然灵敏度降低但仍可使测定结果准确。ρ(Al3+)达到200mg/L时对Ti2+的干扰也成定值,故可加入Al3+达到缓冲的目的。
在消除化学干扰时还应注意,加入稀释剂或保护剂要适量,加入量过少不能起到消除干扰的作用,加入量过大则导致灵敏度下降。因此工作曲线也应加入同量的稀释剂或保护剂,以保证空白和灵敏度相同,从而获得准确的测量结果。
当上述方法不能消除化学干扰时,可采用APDC-MIBK和DDTC-MIBK两个溶剂萃取体系。在分离干扰成分的同时还达到了富集待测元素的目的。
4.2 电离干扰及消除方法
碱金属元素电离电位很低,当火焰温度较高时便会激发或电离,受激发的原子产生背景干扰,而电离原子会导致灵敏度降低。克服电离干扰的方法,一般采用加入消电离剂,即加入比待测元素电离电位更低的元素,或者虽然电离电位较高但加入量大,从而抑制了待测元素的电离。这种加入的金属盐类称为消电离剂。常用的是高纯度的Sr(NO3)2或SrCl2,应注意的是标准系列和试样应同时同量加入,并注意空白的变化及扣除。
4.3 光谱干扰及消除方法
这类干扰是指检测时待测成分的分析线附近存在其它谱线,造成谱线重叠,使测定结果失真。例如Ni元素的分析线232.0nm的两侧还有231.98和232.14nm,这三条谱线对基态原子的吸收灵敏度各不相同,因此在不能将这三条谱线分开时,导致灵敏度下降,线性范围变窄。其它过渡元素Co、Fe也有类似情况。克服光谱干扰的办法是改变谱线通带,即缩小狭缝,或改用无光谱干扰的次灵敏线。
4.4 背景干扰及校正[5]
试液在火焰中产生的背景吸收主要有两类,一是光散射,二是分子吸收,两者均能增加表观吸光度,使测定结果偏高。消除背景干扰的方法是背景校正。通常使用氘灯背景校正器,其基本原理是在灯源旁边引入一个氘灯形成的双光束,转动扇形镜,使氘光与试样发射的光轮流通过狭缝,因氘灯在200~400nm范围内是连续光谱,测量的是背景吸收。扣除后得到的是试样的真实原子吸收,氘灯校正法在波长350nm以下使用效果较佳。
5 原子吸收的定量方法与减免误差[1,3]
原子吸收法的定量方法有很多种,如标准曲线法、标准加入法、内标法以及浓度直读法等,应用最广的是标准曲线法和标准加入法。
5.1 标准曲线法
对于基体成分比较简单的试样,可取一标准系列,用水稀释至一定体积,以试剂空白将仪器调零,然后依次测量各标准溶液的吸光度,给出吸光度-浓度工作曲线。试样在相同的条件下测量吸光度,从工作曲线上求其浓度。也可利用直线的斜率计算试液待测元素的含量。
但是试样含有某些基体成分,如高浓度的酸或盐类或有机溶剂,使试液的表面张力、粘度与标准溶液不同,影响到试样的提升量和雾化效率,因此测得试液与标准溶液的灵敏度不同,不能准确定量。如果基体成分为已知,可进行基体匹配(或称基体修饰),即把已知基体成分加到标准系列中去,然后再进行测量,其影响可消除。
由于具体条件的变化,会影响到工作曲线的斜率。因此工作曲线需每次重新绘制,而且在测定过程中,灵敏度也会发生变化,需要随时核查,并予以校正。
5.2 标准加入法
试样的基体成分经常是无法准确知道的,要用基体匹配的方法就难于实现,在这种情况下用标准加入法比较方便。具体做法,取相同体积的试液3、4份,分别加入标准溶液 O、X1、X2、X3(浓度或体积),用溶剂稀释至一定体积,加入浓度应在良好的线性范围内。然后依次测量各份溶液的吸光度,绘制标准加入法曲线。将加入法直线向左边外延,与横坐标轴相交得Cx即为待测元素的浓度。标准加入法能克服试样中的基体干扰,从而得到精确的测量结果。标准加入法绘制的工作曲线有三种情况,如图1所示:
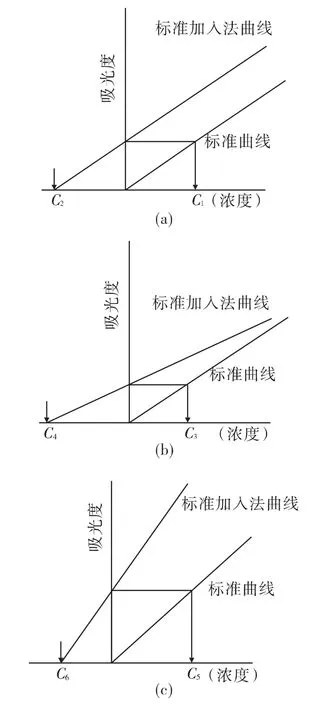
图1 标准加入法检查基体干扰
1)标准加入法曲线与标准曲线平行,说明试样溶液不存在基体干扰,可使用标准曲线法测量;如图1(a)。
2)标准加入法的曲线的斜率低于标准曲线,说明有基体负干扰,如果用标准曲线法定量,会给测量结果带来负误差;如图1(b)。
3)标准加入法曲线的斜率大于标准曲线的斜率,说明试样有基体的正干扰,若用标准曲线法定量,则得偏高的分析结果;如图1(c)。
4)对于2)和3)两种有基体效应的情况应采取标准加入法进行定量。
然而,实际的试样也有可能既存在基体干扰又存在背景干扰,此时标准加入法曲线与标准曲线不平行,应在扣除背景的基础上,用标准加入法定量,或采用上述的消除化学干扰的措施。
5.3 吸光度测量与减免测量误差
1)吸光度的测量应按浓度由低到高的顺序测定标准系列。当测量浓度较高的标准液或试液,需喷入纯水充分冲洗,直至恢复至零才能继续测量,每测10个试样应测量一个标准溶液,检查基线是否漂移,若漂移严重则需重新校零点,前次测量标准后的试样应重测。
2)测量Zn、Na及Ca等元素时往往试样空白较高,此时至少制备3个以上的试样、试剂空白,防止扣除空白时产生误差。
3)吸光度值在0.434附近测量误差最小,最佳吸光度值范围应在0.1~0.6之间。当吸光度值大于1.0时测量误差较大,应适当稀释试样;当小于0.008时应将试液浓缩。
4)原子吸收的灵敏度定义为产生1%吸收信号的试样浓度,即当喷入某元素的溶液进行测量时,得到吸光度值0.004 4时尚不能准确定量,只有空白吸光度值接近于零或很低,且吸光度值大于0.004 4时才能认为是接近灵敏度的数据准确。
5)所用试剂的纯度、器皿的洗涤也会引入误差。宜选用优级纯试剂,在处理试样时必须同时做试剂空白,且试剂加入量应与试样绝对保持一致,并按规定洗涤所有器皿后,置于不受实验室空气污染处。
6)配制标准系列和制备试样时必须逐级稀释,每次最多稀释10倍。切记每次都要加入相应的酸。还应使标准系列与试样的酸度保持一致。
7)试样须用酸进行消解时,消解后的试样应与标准系列保持尽可能相同的酸度。
8)试样的温度与标准系列溶液的温度应保持相同。若从冰箱中取出标准系列未使温度与室温平衡就开始测量,也会产生误差。
此外,仪器条件选择不当也是重要的误差来源。基体效应和背景干扰应按前述方法消除。
6 仪器的日常使用与维护[3,6]
6.1 主 机
1)仪器应安放在无振动、无腐蚀性气体和通风良好的实验室的实验台上,附近不应有强电磁场干扰。室温不应低于5℃,也不应高于30℃。
2)在潮湿的我国南方,应装有除湿机,防止湿气损坏元器件。
3)使用空调的房间,应避免空调机直接对准仪器,以免使测量信号产生波动。
4)气路和冷却水管路严防泄露。
5)仪器长期不用应定期通电,以防元器件受潮损坏。在南方梅雨季节,每周至少通电1h以上。
6)按规定的周期对仪器进行检定,并以基线稳定性、精密度、检出限和灵敏度为主要检定指标。
6.2 空心阴极灯
1)装卸空心阴极灯时,切勿用于触摸顶部的石英窗,否则会沾上汗渍或油污,影响光的透射。使用完毕应及时放入灯盒内妥善保存,防止被沾污、振动。
2)空心阴极灯长期闲置不用,会使发射谱线不纯或不锐,应每隔2个月点燃处理,即在工作电流下处理1h以上。
3)如果空心阴极周围变黑,是长期放置阴极材料变质或使用过高的灯电流阴极金属溅射所致。当无新灯更换时,仍可使用,但灯电流一般比常规电流增加3~4mA,且应使用小通带宽度。
4)通电后空心阴极灯可从阴极发光的颜色大致判断是否正常。充氖灯的负辉光是橙红色,充氩灯是淡蓝紫色。如果长期不使用又未定期电灯处理,则负辉光颜色变淡,呈粉红色或淡蓝白色。此时可将空心阴极灯的阴极与阳极反接处理,阳极一般是棒状,将其接至阴极电源,使用20~30mA电流通电30~60min;通常阴极是由测定元素的纯金属或合金制成的圆形环,将其接至阳极,以100mA通电1~2min,如果是合金阴极通电5min以上。
6.3 光电倍增管
光电倍增管应避免长时间使用高电压,以避免测量信号波动性增加,并避免损坏或疲劳现象的发生,如果开启仪器后不立即测量应关闭高压开关。
1)一般只需使用200V电压,预热20min即可。预热时间过短,暗电流较强,会使测量信号波动性增大。
2)一般仪器使用的光电倍增管只在190~400nm波长范围内灵敏度较高,大多数元素的灵敏波长也在此范围之内。在测定Ca、Sr、K、Na和Li等长波长元素时,可适当提高空心阴极灯的电流,以增大检测信号。
6.4 燃烧器和雾化器
1)燃烧器使用一段时间或测定盐类浓度较高的试样后,会发现火焰呈锯齿状,且在缝隙处有盐类淀积,使测量精度和灵敏度都降低。当积淀盐类较少时,可用滤纸片插入缝中擦净,当积淀物较多时可用刀片轻轻刮去积淀物,但应保持缝隙及其周围的光洁,又不能在金属燃烧器上留下刮痕。当积淀物难以刮去时,可将燃烧器卸下,用10%盐酸或硝酸清洗。在安装时一定使锥形部位连接严密,不能漏气和漏液。
2)每次清洗燃烧器后或更换空心阴极灯时,都应对光路进行简单检查。将划有垂线的方形纸片放在燃烧器上方,使垂线与缝隙垂直,向左右方向移动纸片,观察空心阴极灯的光斑是否在整个缝隙上方通过,否则需对燃烧器或空心阴极灯的位置进行调整。
3)吸液毛细管需更换时,可用一只手掐紧雾化器上的金属毛细管,使之保持水平并固定不会发生进出位移,另一只手更换毛细管。务必使溶液喷嘴同心圆的相对位置不发生变化。
4)雾化器喷嘴前方的撞击球位置对雾化效率影响也较大。若发现雾化效率变差且喷嘴的同心圆部位未曾改变时,应调撞击球前、后、左、右的位置,使喷出的水雾均细且量大,达到最佳状态。在调节喷嘴或撞击球后,将其装入雾化室腔体内,应保证严密并锁紧栓销。
5)雾化室每周应清洗一次。即在试样测定完毕后,关闭仪器主机、空心阴极灯及光电倍增管等电源,喷入10%盐酸或10%硝酸3~5min,然后喷入纯水10min。
6)废液排出管应备有水封,以保持雾化室负压稳定,减少测定误差,保证测量精度。
6.5 空气和燃气
1)空气一般由空气压缩机供给。为了防止空压机的噪声和振动对测量结果产生影响,应远离仪器安放台。压缩空气应有稳压和油污滤除装置,且应经常检查运行是否正常。
2)乙炔钢瓶应远离火源,单独放置在通风良好的位置。应垂直放在专门加工的钢瓶架上并紧固,以防止液态丙酮从气瓶阀中流出损坏气阀和导管。
3)乙炔钢瓶中的气体一般溶解在丙酮中,随着瓶中乙炔压力的降低,进入乙炔火焰中的丙酮浓度会增加,当使用富燃火焰或测定波长低于250nm时,会因混入丙酮而产生较大的测量误差。一般乙炔钢瓶的压力小于0.5MPa时应停止使用。
4)乙炔钢瓶都使用两级减压阀,第一级显示瓶内压力(与钢瓶总阀相连接),第二级显示供给仪器的压力。当测定结束时应首先关闭总阀,使管路中的乙炔流入火焰中燃烧,待火焰熄灭后再关闭二级阀。切勿在管路内残留乙炔气体。
5)燃烧器上方的排风罩用于排出燃烧生成物和试液中的重金属等有害物质。其使用的排风量与排风罩和燃烧器的距离有关,当距离较近时虽然排风效果较好,但会扰动火焰使测量精度降低,故应调节使用的风量。此外,排风罩应使用耐酸材料制作,使用金属排风罩最好涂防锈漆以免对金属测定产生影响;并应经常擦拭灰尘,以免对Ca、Fe及Na等元素的测定产生影响。
6.6 光学系统
1)分光部分多采用光栅及一系列反射和聚光镜组成,密封在仪器的箱壳之内,一般不能随意打开。早期的仪器需更换硅胶以保持干燥,现在的仪器密封严密,只要在通风、干燥的实验室内使用,一般不需用硅胶干燥,因此室内应使用干燥机以保持光学系统的干燥和清洁。
2)在燃烧器的左右两侧都有聚光用的石英透镜,因其暴露在外,容易受尘沾污,且长期使用也会被火焰中测量物质飞溅沾污,从而影响测量灵敏度。此时也可用擦镜纸轻轻擦拭,当沾污严重时可蘸无水乙醇擦拭。
[6]J.A迪安.分析化学手册[M].北京:科学出版社,2003:715-726。
续完
Brief Introduction of Atomic Absorption Technique(Ⅱ)(the end)
QIU Xing-chu1,QIU Shan1,QIU Sheng1,CHU Rong-bang2
TG115.33
B
1001-3849(2012)03-0042-05
2011-05-20
2011-07-26
