隐藏高分子界面及生物界面分子结构的和频振动光谱研究
2012-11-30陈战
陈 战
(密歇根大学化学系,930 North UniversityAve.,AnnArbor,MI 48109,美国)
隐藏高分子界面及生物界面分子结构的和频振动光谱研究
陈 战*
(密歇根大学化学系,930 North UniversityAve.,AnnArbor,MI 48109,美国)
界面的分子结构决定界面的性质.为了以优化界面的结构来改进材料的性质,原位实时地研究界面的分子结构是很重要的.近年来和频振动光谱已发展成为一个很有效及独特的手段来研究隐藏界面的分子结构,例如液/液界面、固/液界面及固/固界面等.这篇综述讨论了和频振动光谱在研究高分子界面及生物界面等复杂界面的分子结构上的应用.具体说来,本文论述了高分子表面在水里的分子结构变化,高分子及模型粘合促进剂硅烷在界面相互作用的分子机理和隐藏的高分子/高分子及高分子/金属界面的结构.另外,此文还将介绍不同二级结构的多肽及几个有代表性的蛋白分子在界面的结构.界面在诸如化学、生物、物理、材料科学及工程和纳米技术等许多领域都很重要.发展一个独特的能原位研究隐藏界面的分子结构的技术会有力地促进这些领域的研究及跨学科研究的发展.
高分子表面和界面;生物界面;多肽;蛋白质;粘合;二级结构;α-螺旋;β-折叠
1 Introduction
Surfaces and interfaces are important in many research fields as well as many aspects of everyday life.Our bodies contain many different interfaces that are vital to our well-being. Interfacial reactions are responsible for protein interaction with cell surfaces,hormone-receptor interactions,and lung function.Modern science has explored and designed surfaces and interfaces for many applications:anti-biofouling surfaces are being researched for medical implants and marine vessels; high temperature resistant surfaces are important for space shuttles;and heterogeneous catalysis,studied by interfacial reactions,is important in industry and environmental preservation.The usefulness of many common items is determined by surface properties:contact lenses must remain wetted;while raincoats are designed to be non-wetting;and coatings are applied to cookware for easy clean-up.Nanoscience and nanotechnology have greatly changed our lives in the past decades. When a material shrinks in size,the ratio of the number of the surface molecules to that of the bulk molecules increases. Therefore surfaces and interfaces are extremely important for nano-materials.
In order to improve properties of surfaces and interfaces such as hydrophobicity,friction,lubrication,adhesion,wearability,interfacial chemical reactivity,and biocompatibility,it is crucial to optimize molecular surface or interfacial structures. The first step to do so is to understand the molecular structures of surfaces and interfaces.However,it is still very challenging to investigate many surfaces and interfaces at the molecule level in situ.
Many different surface sensitive techniques have been developed in the past decades.1,2However many of them require high vacuum to operate,therefore cannot be applied to investigate interfaces involving liquid materials,e.g.,solid/aqueous solution interfaces.In addition,many techniques measure surface/interfacial properties,which cannot provide molecular structural information.Recently,sum frequency generation (SFG)vibrational spectroscopy has been developed into a powerful technique to study various surfaces and interfaces in situ at the molecular level.3-26It has been demonstrated that SFG vibrational spectroscopy is capable of probing interfacial structures of complex surfaces and interfaces,including polymer interfaces and biological interfaces.27-31
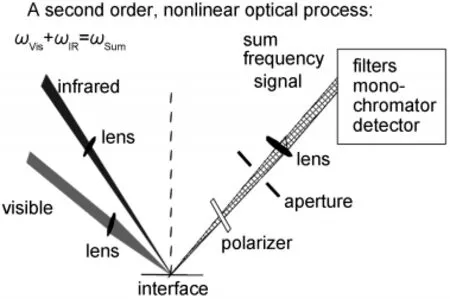
Fig.1 SFG experimental geometryReproduced with permission from reference31.Copyright 2010 Elsevier.
SFG is a process in which two input beams at frequencies ω1and ω2mix in a medium and generate an output beam at the sum frequency ω=ω1+ω2(Fig.1).3-31The selection rule for this three-photon process indicates that SFG is only allowed in media without inversion symmetry(under the electric-dipole approximation).3-31Most bulk materials possess inversion symmetry,thus do not generate SFG signal.At surfaces or interfaces where the inversion symmetry of the bulk is broken,an SFG signal can be generated.Both experimental evidence and calculations show that SFG is submonolayer surface sensitive.For IR-visible SFG,the IR input beam,ω1,is frequency tunable. When ω1is in resonance with a vibrational mode of a functional group on a surface or at an interface,the SFG signal intensity can be enhanced.The intensity of the SFG signal vs ω1can produce the vibrational spectra of the surface species.Various surface or interfacial phenomena can be observed using SFG. For examples,the development of hydrogen bonds can be detected by peak shifts of certain functional groups,while the formation of chemical bonds can be deduced from the appearance of new vibrational modes and disappearance of existing modes in the SFG spectra.The alignment,orientation,and orientation distribution(or ordering)of surface functional groups can be deduced by examining SFG spectra collected using different polarization combinations of input and output beams,and the absolute peak intensities.3-31
Two SFG systems are available and the third system is being built in our lab at the University of Michigan.Each setup is comprised of four components:a pico-second Nd:YAG laser,a harmonics unit with two KD*P crystals,an optical parametric generation(OPG)/optical parametric amplification(OPA),and difference frequency generation(DFG)system,and a detection system.32,33The visible beam is generated by frequency-doubling the fundamental output pulses,of 20 ps pulse width, from the laser.The IR beam can be tuned from 650 to 4300 cm-1(15.4 to 2.3 μm).The incident angles of the visible and the IR input beams are 60°and 54°versus the surface normal, respectively,and the diameters of both beams at the surface/ interface are about 500 μm.SFG spectra with different polarization combinations including ssp(s:polarized SFG output,s: polarized visible input,and p:polarized IR input),ppp,and sps can be collected.
In this paper,I will summarize the recent progress of SFG studies on polymer interfaces and biological interfaces,focusing on the research outcomes from our lab at the University of Michigan.Particularly,molecular surface structural changes of various polymers in water,polymer-silane interactions,as well as structures of buried polymer/polymer and polymer/metal interfaces will be presented.In addition,interfacial structures of peptides with varied secondary structures and several representative proteins will also be discussed.SFG is a powerful and unique analytical technique which is revolutionizing the surface science.
2 Polymer interfaces
2.1 Surface restructuring in water
Polymer materials have many applications in the aqueous environments while their surface structures are important for their functions.For example,polymers such as silicones or polyurethanes are routinely implanted into human bodies,coming into contact with blood and other tissue,while marine antibiofouling polymer coatings are used in ocean.34-39Surfaces of polymer membranes for biomolecule separations are exposed to biomolecule solutions when in use.To ensure the desired performance of these polymers,it is important to optimize the polymer surface structures in aqueous environments.The first step for this purpose is to investigate these polymer surface structures in situ while in contact with aqueous solutions.Due to a lack of appropriate technique,it is difficult to examine polymer/solution or polymer/water interfaces.Traditionally, water contact angle experiments were used to monitor the surface structural changes of polymers after contact with water,40-43but they do not provide molecular level structural information.Freeze-drying X-ray photoelectron spectroscopy (XPS)has been used,but its sample preparation is complicated and it cannot provide orientation information of functional groups on the surface.44SFG has been shown to be powerful and unique in studying solid/liquid interfaces.SFG can provide molecular structural information of polymer/liquid interfaces in situ in real time.27,31
2.1.1 Different polymer surfaces exhibiting varied
surface structural changes in water
It was believed that polymer surfaces could exhibit restructuring behavior in water.Because different polymers may exhibit varied surface restructuring behaviors in water,it is difficult to predict polymer surface structures in water from the results obtained from the experiments performed in air or in high vacuum.It is necessary to characterize polymer surfaces in water in situ.
The first application of SFG to study polymer surfaces in water was published in 1997.45SFG was used to monitor the surface structure of a polyurethane with poly(dimethyl siloxane) (PDMS)end groups in water.45The surface restructuring behavior in water was clearly observed.The results indicated that the hydrophilic polyurethane backbone tended to segregate more to the surface in water while the hydrophobic PDMS end groups retreated to the bulk.This early research has demonstrated the feasibility to investigate surface restructuring behavior of polymer materials in water in situ using SFG.Since then, SFG has been applied to study surface structures of many different polymers in liquid environments including water.
In our lab at the University of Michigan,we extensively investigated surface structures of different polymethacrylates with varied side chain lengths.32,33,46-50Our results showed that different surface restructuring behaviors,e.g.,reversible side chain orientation changes and irreversible backbone changes of polymer surfaces in water,can be observed using SFG.32,48This provides direct experimental evidence that different polymers do exhibit varied surface restructuring behaviors in water.In more detail,it was found that poly(methyl methacrylate) (PMMA)surface is dominated by the ester methyl groups, which do not show noticeable changes in water.32This is due to the fact that the ester methyl groups are more hydrophilic compared to the normal methyl groups,therefore they do not need to reorient in water when interacting with the water molecules. Also the PMMA glass transition temperature is high;the surface can be quite rigid,similar to the bulk.Therefore there is no surface restructuring behavior occurred in water.SFG spectra indicated that poly(n-butyl methacrylate)(PBMA)(Fig.2)46and poly(n-hexyl methacrylate)(PHMA)surfaces are dominated by side chain methyl groups.32,48Upon contacting with water,these surface dominating methyl groups tilt more towards the surface.Such surface structural changes are reversible.The SFG spectra recovered after removing the surfaces from water and exposing them to air again.For longer side chain polymethacrylates,such as poly(n-octyl methacrylate)(POMA) and poly(n-lauryl methacrylate)(PLMA),surface structures be-come random in water,evidenced by the disappearance of the SFG signal.32Such changes are irreversible.This is because POMA and PLMA have very low glass transition temperatures;the surfaces can be quite mobile,leading to irreversible surface restructuring in water.For poly(n-octadecyl methacrylate)(PODMA),the side chain is even longer.Because the PODMA surface is semi-crystalline,it exhibits a reversible surface restructuring behavior in water.48Using polymethacrylates as examples,we clearly demonstrated that different polymer surfaces exhibit varied surface restructuring behaviors in water.
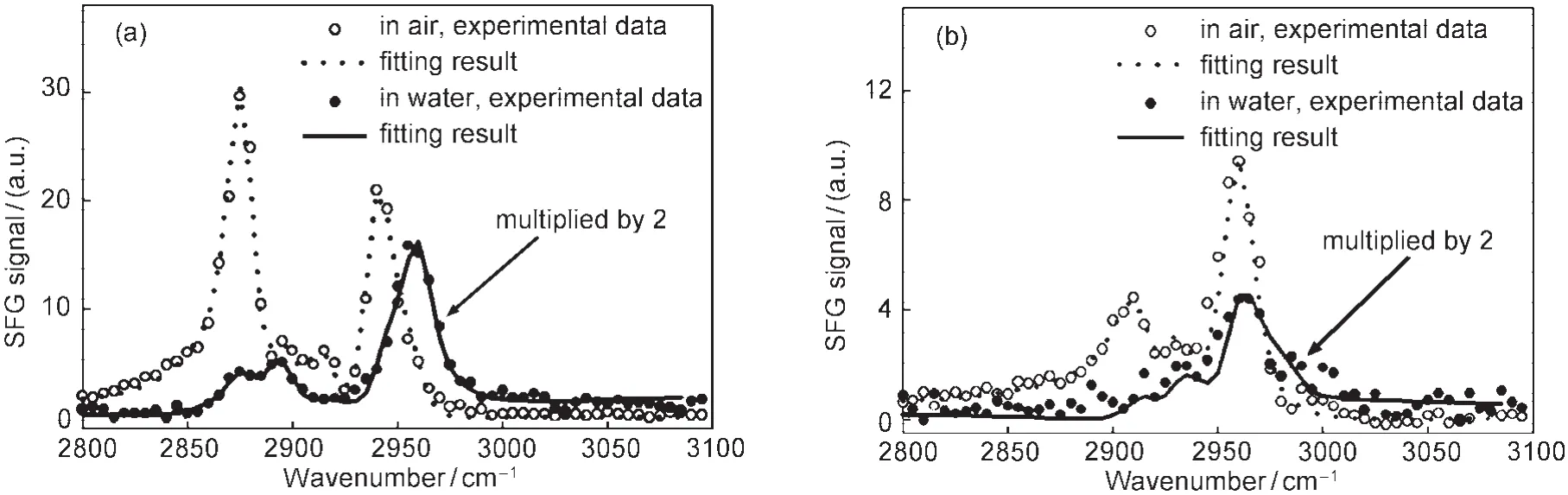
Fig.2 SFG spectra of PBMAin air and water for(a)ssp and(b)sps polarization combinationsReproduced with permission from reference46.Copyright 2002Am.Chem.Soc.
2.1.2 Quantitative study of polymer surface structure
Polymer surfaces can be quite complicated therefore it is difficult to describe polymer surfaces quantitatively.We believe that if we can quantify the surface coverage of various functional groups,their average orientations and orientation distributions,we then should be able to quantitatively describe a polymer surface using these parameters.
We developed a methodology by using SFG to quantify the orientation distribution of dominating methyl groups on a PBMA surface in air and in water.46In order to do so,we have collected SFG spectra using different polarization combinations and calibrated the absolute SFG spectral intensity.We deduced the orientation distribution of methyl groups using a Gaussian function as well as a maximum entropy trial function.46Both functions lead to similar results,indicating that in this case a Gaussian distribution is a good approximation for the orientation distribution.
It is interesting to see that the deduced orientation distribution for methyl groups on the PBMA surface in air is broader, with a small average orientation angle vs the surface normal (stand up more).46But in water,the PBMA surface methyl groups have a narrower orientation distribution and the average orientation angle is larger(lie down more).In air,the methyl groups have more freedom,resulting in a larger orientation distribution.The methyl groups are hydrophobic,and air is also hydrophobic,therefore methyl groups can stand up in the air on the PBMA surface.In water,due to the unfavorable interactions between water and the methyl groups,the methyl groups were pushed down by water molecules,leading to a larger average tilt angle.However in water the orientation distribution is narrower,indicating that the polymer surface is more ordered. In addition to PBMA,the orientation distributions of methyl groups on a series of polymethacrylates with different side chain lengths in air and in water have also been deduced using similar approaches(Fig.3).48
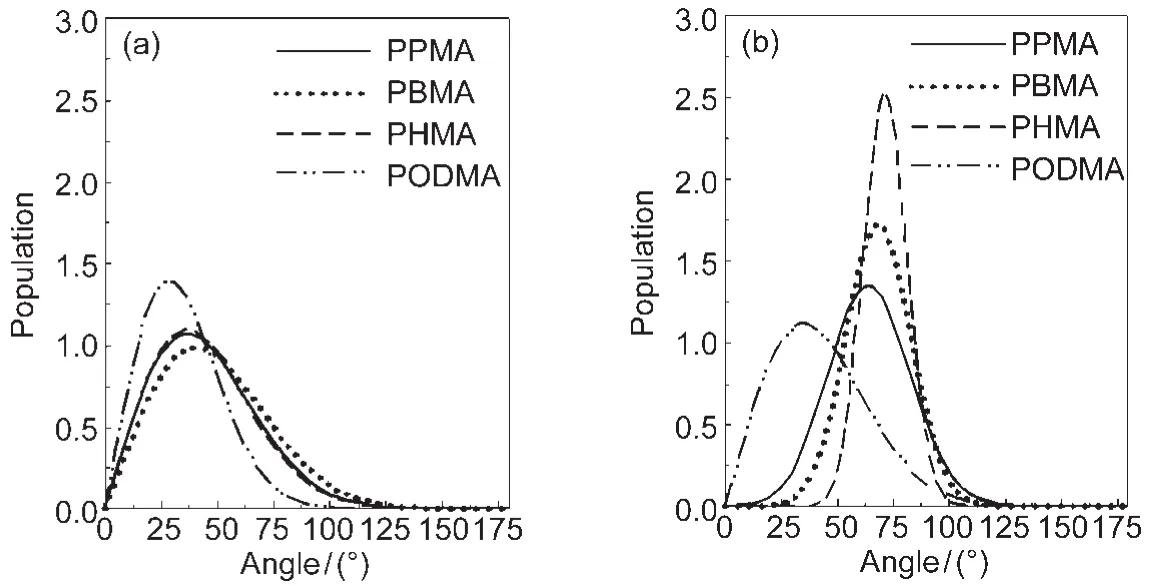
Fig.3 Orientation distributions deduced for the side chain CH3groups on the surfaces of poly(n-propyl methacrylate)(PPMA),poly(n-butyl methacrylate)(PBMA),poly(n-hexyl methacrylate)(PHMA),and poly(n-octadecyl methacrylate)(PODMA)in (a)air and in(b)waterReproduced with permission from reference48.Copyright 2006Am.Chem.Soc.
To study polymer surfaces in air and in water,polymer films were deposited onto fused silica or CaF2substrates using spin coating.In the most situations,SFG signals collected from the polymer/fused silica or polymer/CaF2interfaces are quite weak and can be ignored.Since the SFG signals collected from the polymer/water interface can be quite weak as well,sometimes it is necessary to consider the signal from the buried polymer/ substrate interface.In the PBMA/water interface studies,we later found that weak SFG signals can also be detected from the PBMA/fused silica interface.49We then developed a systematic means to separate the SFG signals contributed from the PBMA/substrate interface and the PBMA/water interface by varying the PBMA film thickness,probing the molecular structures at the two buried interfaces simultaneously.49Using a similar approach,we also studied a tri-block copolymer developed for marine anti-biofouling coatings.51Using SFG,we demonstrated that the fluorinated groups in the polymer can be present on the surface in water.These hydrophobic fluorinated groups were brought to the surface in water by their neighboring hydrophilic ethylene glycol groups.51
2.1.3 SFG studies on silicone
Silicone such as poly(dimethyl siloxane)(PDMS)has been widely used as biomaterials and marine anti-fouling coatings.36-39Since these PDMS materials are used in the aqueous environment,their surface structures should be characterized in water in situ.SFG has been applied to elucidate the molecular surface structures of several model PDMS materials in air and in water.52It was found that in both environments these PDMS surfaces are dominated by Si―CH3groups,but the orientations of such Si―CH3groups on the surface are different in air and in water.
In addition to the model PDMS materials,SFG has also been applied to investigate PDMS materials incorporated with biocides.Coupling biocides into polymer coatings may be another effective means to prevent marine biofouling from occurring.PDMS is being developed as a promising fouling release polymer coating for marine vessels.36,37Incorporating biocides into PDMS may lead to polymer coatings which can have both anti-fouling and fouling release characteristics.For example, the quaternary ammonium salt(QAS)was incorporated into PDMS and we have investigated the effect of tethered QAS chain length(the lengths of R1and R2in Fig.4)53on PDMS surface structures and their antifouling performance.53The chemical compositions of the compounds we investigated are shown in Table 1.53
Since the application for these PDMS materials is for an aqueous environment,SFG spectra were collected from the surfaces contacted to water(Fig.5).53The SFG spectra for the samples with the chain length of R2being 3(labeled as R2=3 for simplicity)are quite different from the spectra for the samples with the chain length of R2being 11(labeled as R2=11 for simplicity).We believe that they adopt different surface structures in the aqueous environment.After carefully analyzing these spectra,we can deduce surface structures of PDMS incorporated with QAS with different R1and R2chains.The schematics of such surface structures are displayed in Fig.6.53
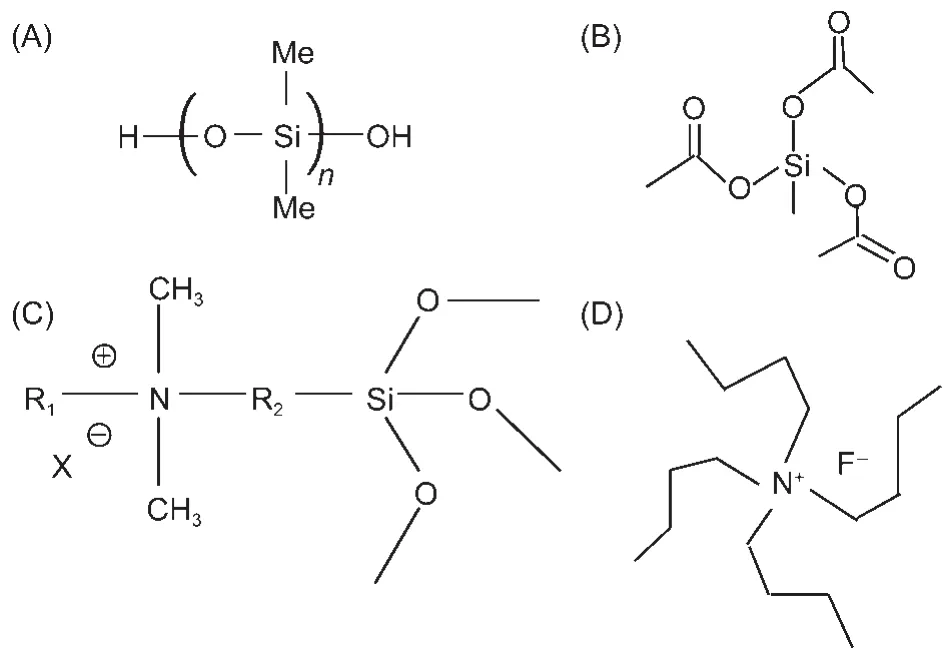
Fig.4 Molecular formulas of the materials used to prepare PDMS incorporated with QAS(A)silanol terminated PDMS,(B)crosslinker,methyltriacetoxysilane, (C)QAS,(D)catalyst,tetrabutylammonium fluoride(TBAF);Reproduced with permission from reference53.Copyright 2010Am.Chem.Soc.
To correlate the molecular structures of PDMS incorporated with QAS to the antifouling behavior of these materials,we also tested the antifouling activities of various samples towards different microorganisms(Table 2)53.For R2=3,surfaces in water were covered by R1.The long R1chain in QAS moieties resulted in a more hydrophobic surface.Since QAS moieties provide effective antimicrobial activity through a contact mecha-nism,the microbial cells can be adsorbed onto solid surfaces by electrostatic and/or hydrophobic interactions.If the R1chain is sufficiently long and flexible to penetrate the adsorbed bacterial cell walls,the QASs can disrupt the cell membrane to cause cell death.The surfaces of A3 and A5 are more hydrophobic than A1,leading to more active antifouling performance thanA1.
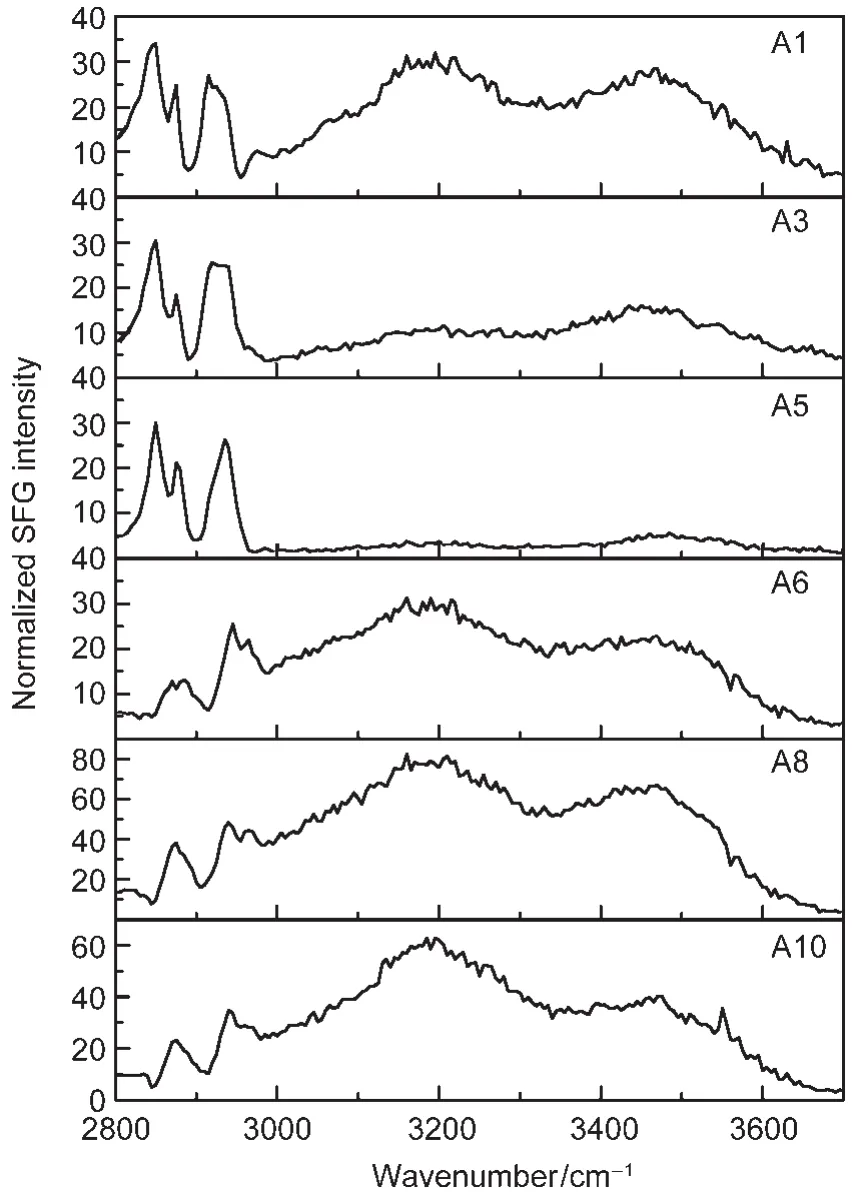
Fig.5 SFG spectra(ssp)of various QAS incorporating PDMS samples collected in waterReproduced with permission from reference53.Copyright 2010Am.Chem.Soc.
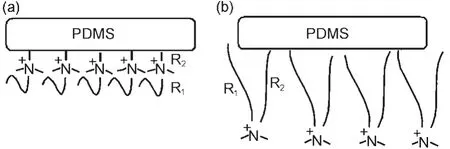
Fig.6 Schematics of molecular surface structures of the QAS incorporating PDMS samples with R2=3 in water(a)and with R2=11 in water(b)Reproduced with permission from reference53.Copyright 2010Am.Chem.Soc.
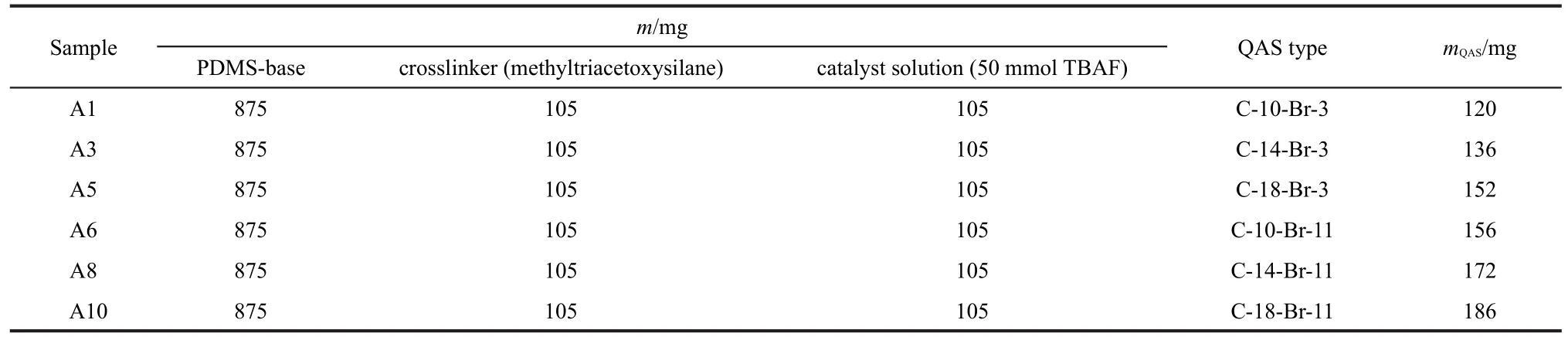
Table 1 Formulations based on silanol-PDMS and QASs
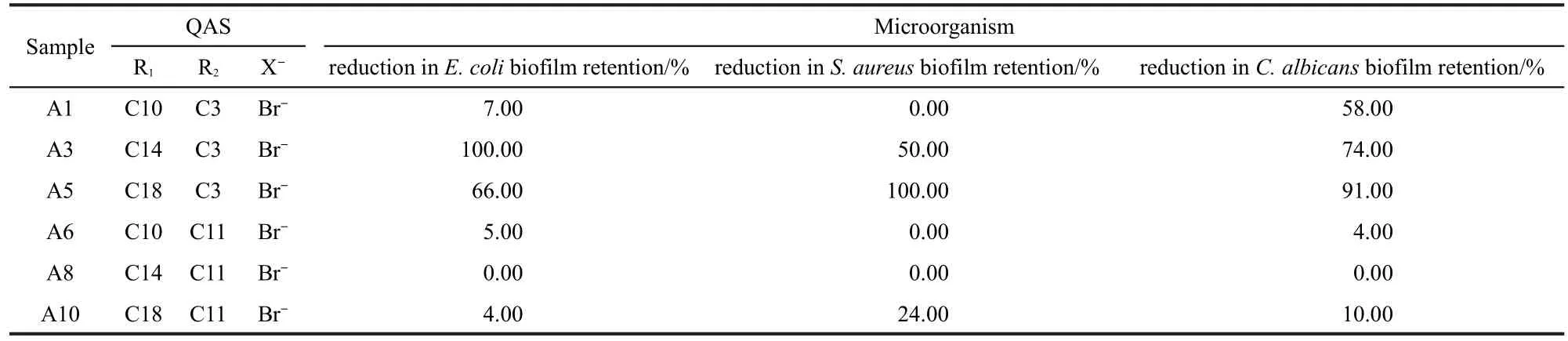
Table 2 Activities of various samples towards different microorganisms
For R2=11,the coating surfaces restructure in water and the nitrogen atoms in the QAS moieties segregate to the surface, so the coating surfaces become hydrophilic.Hydrophilic surfaces exhibit high resistance to nonspecific protein and microorganism adsorption.Thus the PDMS with QAS of R2=11 was ineffective at reducing the biofilm retention of E.coli,S.aureus, and C.albicans because the interaction between the coating surfaces and the water molecules can resist the adsorption of microbial cells and then the QASs do not have the opportunity to contact microbial cells and kill them.On the other hand,the interaction of coating surface with the functional groups on microorganisms involves a balance between electrostatic and hydrophilic interactions.It is not unexpected that the coatings of A6 and A8 show somewhat active antifouling performance at reducing the biofilm retention of C.lytica since its outer cell membrane has different compositions.Here,electrostatic forces will play a dominant role in driving the adsorption of C.lytica on the coating surfaces ofA6 andA8.
It has been extensively demonstrated that zwitterionic surfaces can effectively minimize biofouling to occur.54-56We therefore incorporated zwitterionic functional groups into PDMS materials to develop surfaces with both antifouling and foulingrelease properties.We successfully coupled a zwitterionic group into PDMS using UV irradiation,confirmed by contact angle measurements and X-ray photoelectron spectroscopy (XPS).57According to the AFM and ATR-FTIR studies,much less protein molecules were adsorbed to such surfaces compared to the PDMS surface without incorporating the zwitterionic group.This demonstrated that it is feasible to couple zwitterionic groups to PDMS surfaces using UV irradiation,and such materials exhibit better protein-resistant properties,which may possess both antifouling and fouling-release properties. Surface structures of PDMS samples incorporated with zwitterionic groups in air and in water were characterized using SFG.57
2.2 Polymer/silane interactions
Silane molecules are widely used as adhesion promoters to enhance adhesion of interfaces involving polymer materials. We have previously investigated interactions between polymers and various silanes.Our studies indicated that silanes can adopt varied conformations or orientations at different polymer interfaces,58can diffuse through polymer interfaces,59,60and can form interfacial hydrogen bonding.61,62Here we will focus our studies on the molecular interactions between an epoxy silane (3-glycidoxypropyl)trimethoxysilane(γ-GPS)and a representative polymer poly(ethylene terephthalate)(PET).Both materials are widely used in industry.
It was observed that the mixture of γ-GPS and an oligomericsiloxane(MVS)better promotes adhesion at the PDMS/PET interface compared to the use of γ-GPS only,therefore the mixture of γ-GPS and MVS is widely used as an adhesion promoting system in industry.63,64The molecular formulas for these two materials as well as PET are shown in Fig.7.63To understand why the mixture better promotes the adhesion,we first investigated the molecular interactions between PET and γ-GPS as well as those between PET and the γ-GPS-MVS mixture (Fig.8).63We used PET with deuterated aliphatic groups (d4-PET)to avoid spectral confusion between the polymer and the silane.The silane methoxy headgroups have net orientational order at the d4-PET/γ-GPS interface,evidenced by a weak 2835 cm-1signal due to the symmetric C―H stretching of the methoxy headgroups.The SFG spectrum collected from the in-terface between d4-PET and a γ-GPS and MVS(1:1 ratio,w/w) mixture(we will call this 1:1 mixture the“silane adhesion promoting mixture”or“SAPM”throughout this paper)immediately after d4-PET contacted with the SAPM contained no resolvable peaks.The 2835 cm-1peak appeared after about 10 min and its intensity remained stable thereafter.This implies that initially either the γ-GPS molecules were not at the interface,they were present at the interface but with no net orientational order,or they lay flat.With time the silane molecules segregate to the d4-PET/SAPM interface with a net orientational order.The larger peak intensity at the d4-PET/SAPM interface implies that methoxy groups had a relatively narrower orientation distribution and the γ-GPS molecules were more ordered towards the surface normal at this interface than at the d4-PET/silane interface.
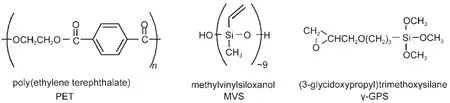
Fig.7 Molecular formulas for PET,MVS,and γ-GPSReproduced with permission from reference63.Copyright 2006Am.Chem.Soc.
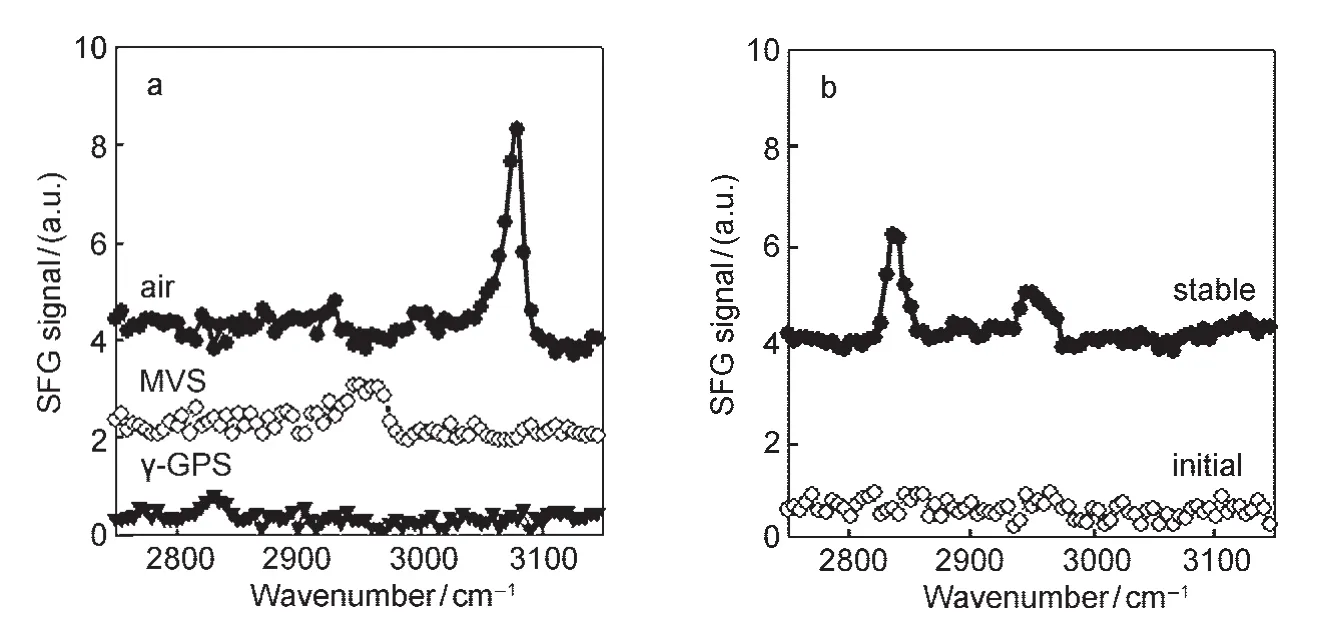
Fig.8 (a)SFG spectra collected from the interfaces between d4-PET and air,MVS,or γ-GPS;(b)SFG spectra collected from the d4-PET/1:1 SAPM interfaceReproduced with permission from reference63.Copyright 2006Am.Chem.Soc.
SFG spectra were also collected from the d4-PET/silane mixture interfaces in which the γ-GPS:MVS mass ratio was varied. The SFG spectra from the interfaces in which the silane:siloxane mass ratios were 1:1,2:3,and 1:2 were dominated by the 2835 cm-1peak.The silane:siloxane mass ratios that provided the greater peak intensities at the d4-PET/SAPM interfaces are similar in magnitude to the mass ratio range claimed to be effective for adhesion promotion in PDMS.65,66Thus,interfacial structure,as deduced by SFG,can prove to be important in determining adhesive strength.However,to understand the direct relation between interfacial structure and adhesion,we need to study interfacial behavior of adhesion promoters at polymer/ polymer interfaces(see below),not only the polymer/silane or polymer/silane-siloxane mixture interfaces.
Since the adhesion promoters were used at polymer interfaces to enhance adhesion,we then studied molecular structures of the γ-GPS and SAPM at the PET/PDMS(before and after curing)interfaces.SFG spectra of d4-PET in contact with uncured PDMS with the γ-GPS and its mixture with MVS were collected(Fig.9a).64The silane concentration is 1.5%(mass fraction)in PDMS and the silane-MVS mixture is 3.0%.Only the SFG spectrum of d4-PET surface in contact with uncured silicone elastomer with 3%SAPM exhibited a peak at 2835 cm-1.64Therefore,when mixed with MVS,the γ-GPS methoxy groups can segregate to the polymer interface with order,when mixed in uncured silicone.For pure γ-GPS,there was no evidence of the silane molecules segregating to the d4-PET interface with order(Fig.9a).This study showed that not only interactions between the d4-PET and silanes but also interactions between the silicone and the silanes affect the ability of silanes to segregate with order at the polymer interface,a key step in adhesion promotion.
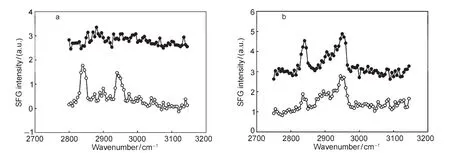
Fig.9 (a)SFG spectra ofd4-PET in contact with uncured silicone with 1.5%γ-GPS(filled)and with 3%1:1 γ-GPS/MVS mixture(open); (b)SFG spectra ofd4-PET/cured silicone interfaces with 1.5%γ-GPS(filled)and with 3%1:1 γ-GPS/MVS mixture(open) Reproduced with permission from reference64.Copyright 2009 Elsevier.
SFG spectra were acquired from d4-PET/cured silicone inter-faces with γ-GPS or its mixture(incorporated into the silicone matrix prior to curing)after curing for 1 h at 150°C(Fig.9b). Both SFG spectra of d4-PET/cured silicone elastomer with 1.5%γ-GPS interface and of d4-PET/cured silicone elastomer with 3%1:1 γ-GPS-MVS mixture interface have the 2835 cm-1peak.64However,the intensity of the γ-GPS at the d4-PET/ cured silicone elastomer(with 1.5%silane)interface is stronger compared to that when 3%SAPM was used.This was the opposite of the relative intensities for the spectra of d4-PET in contact with the liquid SAPM or the uncured silicone elastomer with SAPM.64This may be because the silicone sample with SAPM was cured above the cohesive failure temperature (TCF),which induced a chemical reaction or diffusion of the silane at the polymer interface.This study clearly demonstrates that the MVS molecules interact with γ-GPS at the buried interfaces and affect adhesion.
2.3 Polymer/polymer and polymer/metal interfaces
SFG has been applied to investigate molecular structures of buried interfaces involving polymer materials using different experimental geometry.31,67-77Before we studied the PET/ PDMS interfaces as discussed above,we have already directly probed buried polymer/polymer interfaces,using the buried polystyrene(PS)/PBMA interface as an example(Fig.10).67A series of experiments were performed to ensure that the SFG spectra we collected were contributed by the polymer/polymer interface,not from the PS/substrate or PBMA/air interfaces.67It was found that orientation of side chain methyl groups at the buried PBMA/PS interface is between those at the PBMA/air and PBMA/water interfaces.As discussed above,the orientations of methyl groups on the PBMA surface in air and in water have been quantified.In air,the methyl groups on the PBMA surface have greater freedom and they tend to extend into the air,leading to a small average orientation angle versus the surface normal.46In water,the strong repulsion between the methyl groups and water molecules directs the methyl groups away from the surface normal,resulting in a larger average orientation angle.46At the buried PBMA/PS interface,the methyl groups interact with phenyl groups of PS,and the repulsion must be weaker compared to that with water.The repulsion force also directs the phenyl groups away from the interface normal more compared to the PS/air interface,shown by different aromatic C―H stretching signals at the PS/PBMA and PS/ air interfaces.67
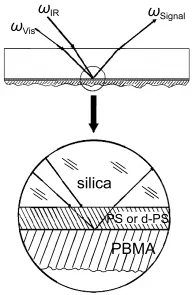
Fig.10 SFG sample geometry for detecting buried polymer/ polymer interfacesReproduced with permission from reference67.Copyright 2002Am.Chem.Soc.
Polymer/metal interfaces are important in many applications including microelectronics and anti-corrosion coatings.We developed two methods to investigate molecular structures of buried polymer/metal interfaces in situ using SFG.We demonstrated our first method by using a PMMA/silver(Ag)interface as a model for polymer/metal interfaces.68To elucidate the structure of the PMMA/Ag interface,SFG spectra were collected from PMMA films with different thicknesses deposited on Ag substrates(Fig.11).68Such SFG signals are contributed from the interference of signals generated from the PMMA/air and PMMA/Ag interfaces,along with some nonresonant contributions.The dominating contribution comes from the PMMA/air interface.When the PMMAfilm thickness is changed,the interference effects also vary.SFG signals from the buried PMMA/ Ag interface have been successfully deconvoluted from the detected interference results by fitting the SFG spectra using the contributions from the PMMA/air and PMMA/Ag interfaces. The molecular structure of the PMMA/Ag interface can be inferred from the deconvoluted SFG signal from this buried interface.68As the PMMA/air interface,the ester methyl groups are dominating the PMMA/Ag interface.Quantitative analysis indicates that they point away from the Ag surface with a large tilt angle.68
The first method discussed above is complicated because SFG spectra need to be collected from polymer samples with different thicknesses.In addition,these spectra need to be fitted using the same set of parameters simultaneously(only varying the film thickness).To overcome these difficulties,we recently developed a second method to study polymer/metal interfaces in situ using SFG.This method can be used to directly probe the buried polymer/metal interface,using the poly(methyl acrylate)(PMA)/silver(Ag)interface as a model.Instead of using polymer films in air with different thicknesses,we col-lected SFG signal from a thin PMA film sandwiched between a fused silica substrate and a silver surface.69Different from what was observed above,the detected SFG signal intensity does not depend on the PMA film thickness(Fig.12).69It was shown that SFG signals were solely contributed by the PMA/Ag interface,no SFG signal was generated by the polymer/fused silica interface(due to the disordered polymer structure at this interface).Further quantitative studies indicated that the ester methyl groups at the PMA/Ag interface tilt away from the Ag surface.69
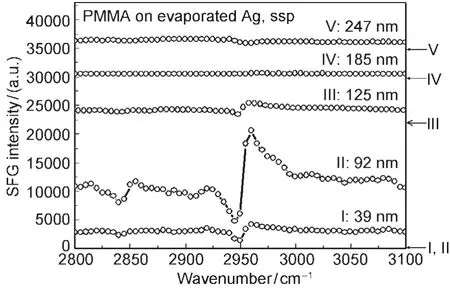
Fig.11 The ssp SFG spectra of PMMAfilms with different thicknesses on the silver substratesThe arrows on the right indicate the baseline for each spectrum.Reproduced with permission from reference68.Copyright 2008Am.Chem.Soc.
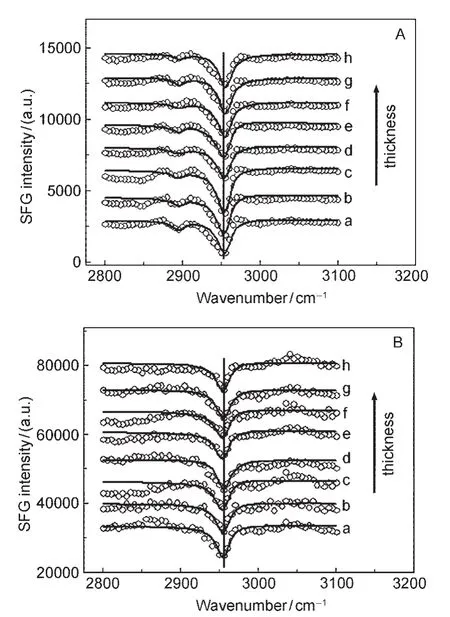
Fig.12 SFG spectra of the PMAthin films sandwiched between the silica window substrate andAg surfaceThe PMAthin film thicknesses of samples a,b,c,d,e,f,g,h are 19,25,41,67, 89,111,136,153 nm,respectively;the spectra have been offset for clarity. A:ssp spectra;B:ppp spectra.Reproduced with permission from reference69. Copyright 2009Am.Chem.Soc.
3 Biological interfaces
It is important to understand molecular structures of biological molecules such as peptides and proteins at interfaces.For example,when a biomaterial is implanted in a human beingʹs body,the first body reaction is protein adsorption.The adsorbed protein structure will influence later body reactions and determine whether the material can be accepted or not.Therefore it is important to study the adsorbed protein structures at the implant/body interface.Similarly,the first step for marine biofouling to occur is the interactions between the adhesive proteins generated from marine organisms and the ship coating surface.The studies on interfacial structures of these adhesive proteins are needed for developing improved coating materials. It is important to study peptides at interfaces as well.Drug resistance has been developed in many bacteria against traditional antibiotics;to overcome this problem,a variety of antimicrobial peptides which can disrupt bacterial cell membranes have been researched to replace conventional antibiotics.The conformation and orientation of these antimicrobial peptides in different cell membranes play important roles in their activity and selectivity,and should be investigated.Elucidation of molecular structures of peptides and proteins at interfaces provides a foundation to understand molecular mechanisms of biocompatibility of biomaterials and marine biofouling,and antimicrobial peptideʹs potency and selectivity.
It is difficult to determine molecular structures of proteins and peptides at interfaces because these structures(especially for proteins)can be very complicated.If the orientations of various secondary structures in a protein at the interface can be determined,a rough structure of the protein may be deduced. SFG has been applied to study proteins and peptides at various interfaces.78-93In the recent years,we developed a systematic approach to determine conformation and orientation of various secondary structures at interfaces using polarized SFG,supplemented by attenuated total reflectance Fourier transform infrared(ATR-FTIR)spectroscopy.28,29,94-99We focused on the studies of interfacial orientations of peptides with certain secondary structures,and then expanded such studies to more complicated peptides and proteins.
3.1 Studies on α-helices
It has been widely demonstrated in linear vibrational spectroscopic studies that different protein secondary structures generate amide I signals with varied peak centers.100-102We demonstrated the feasibility of detecting SFG amide I signals from various proteins at the solid/liquid interfaces in situ.103We also showed that peptides with different secondary structures generate SFG amide I signals with varied peak centers,similar to what was observed by linear vibrational spectroscopy.104We then systematically developed the methodology to characterize orientation of helical structures at interfaces using SFG spectra collected using different polarization combinations.94,96-98For α-helices,we have the following equations.
For theAmode:
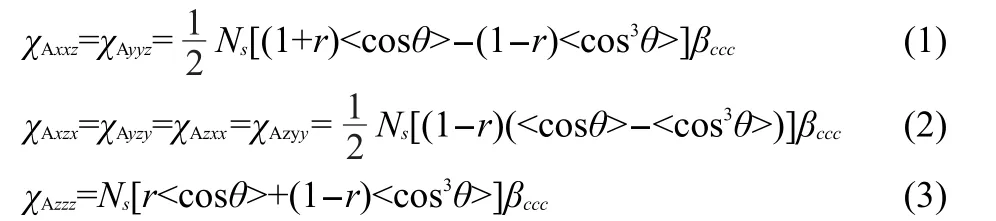
where r=βaac/βccc
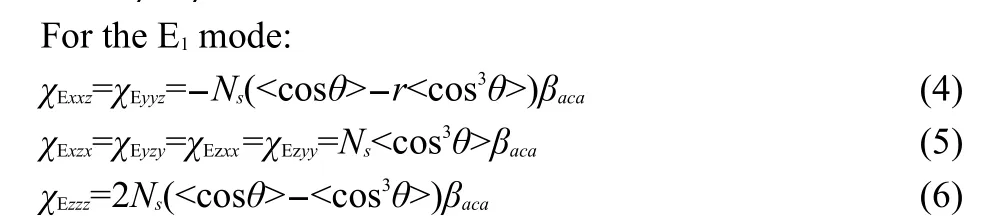
where Nsis the number density of α-helix units,the notation“<>”indicates the average value,and θ is the angle between the principal helical axis and the surface normal.When assuming that θ has a delta-distribution,<cosθ>=cosθ.The χijkterms are second order nonlinear optical susceptibility tensor components,which can be measured using different polarization combinations of the input and output beams in the SFG experiments.The βlmnterms are hyperpolarizability tensor components of the α-helix,which are known(or can be calculated94).
For the α-helix,the A and E1 modes have very similar peak centers,which cannot be differentiated using our SFG spectrometer.Therefore we used the sum of the two modes to perform data analysis.From the above formulas,it is clear that the different second order nonlinear susceptibility components are connected with differenthyperpolarizability components through the average orientation.Since different hyperpolarizability components can be obtained for α-helices94,from the measured second order nonlinear optical susceptibility components,we can deduce α-helix orientation.94
We then applied this data analysis methodology to study orientations of various α-helical peptides at interfaces.Here we started the discussion from the SFG studies on a widely studied antimicrobial peptide magainin 2.
3.1.1 Magainin 2
Isolated from the African clawed frog Xenopuslaevis,magainins have been shown to have antimicrobial activity and have been extensively studied using a variety of analytical techniques.105-112SFG and ATR-FTIR have been applied to investigate molecular interactions of magainin 2 with model cell membranes including 1-Palmitoyl-2-Oleoyl-sn-Glycero-3-[Phospho-rac-(1-glycerol)](POPG)bilayer and 1-Palmitoyl-2-Oleoyl-sn-Glycero-3-Phosphocholine (POPC)bilayer.113The POPG and POPC lipid bilayers were used to model the bacterial and mammalian cell membranes,respectively.SFG results indicated that magainin 2 lies down on the POPG lipid bilayer surface at low solution concentrations,around 200 nmol·L-1.At a higher concentration of 800 nmol·L-1of magainin 2,it was found by both SFG and ATR-FTIR that magainin 2 molecules insert into the POPG bilayer and adopt a transmembrane orientation with an angle of about 20°from the POPG bilayer normal.113For the POPC bilayer,no SFG signal can be detected at 800 nmol·L-1magainin 2 concentration.At a much higher peptide concentration of 2.0 μmol·L-1,SFG studies indicated that magainin 2 molecules adopt an orientation nearly parallel to the bilayer surface,with an orientation angle of 75°from the surface normal,and no ATR-FTIR signal was detected.This shows that SFG has a much better detection limit than ATR-FTIR and can therefore be applied to study interfacial molecules with much lower surface coverage.113The results obtained from this magainin 2 orientation study are well correlated to the theoretical simulation results.114Further investigation of the lipid bilayer SFG signals supports the proposed toroidal pore model for the antimicrobial activity of magainin 2.113
3.1.2 MSI-78
In order to improve the antimicrobial activity and selectivity of AMPs,many other AMPs have been investigated in addition to magainin 2.It was found that a magainin analogue,MSI-78, exhibits improved performance compared to magainin 2.In our group,SFG was also applied to determine the membrane orientation of MSI-78 in various model cell membranes.115In addition to the solid substrate supported POPG bilayer used above,we also used single 1,2-Dipalmitoyl-an-Glycero-3-[Phospho-rac-(1-glycerol)](DPPG)bilayer and 1,2-Dipalmitoyl-an-Glycero-3-Phosphocholine(DPPC)bilayer.Similar to magainin 2,MSI-78 adopts an α-helical structure in the cell membranes.For the DPPG bilayer,the SFG results indicate that the orientation of MSI-78 associated with the bilayer depends on the peptide concentration(Fig.13).115At low MSI-78 concentrations,e.g.,400 nmol·L-1,the helical MSI-78 molecules tilt at the bilayer surface with~70°deviation from the bilayer normal.When the concentration is increased to 600 nmol·L-1,MSI-78 molecules stand up more,changing the orientation to make a 25°tilt from the lipid bilayer normal.At an even higher peptide concentration,multiple orientations of MSI-78 were observed,in agreement with toroidal-type pore formation as reported from a previous solid-state NMR study.116In contrary,no interaction between MSI-78 and a zwitterionic DPPC bilayer(model for mammalian cell membrane) was observed even at a much higher peptide concentration (~12000 nmol·L-1),demonstrating an excellent selectivity of MSI-78.Interestingly,the peptide exhibits a concentrationdependent membrane orientation in lamellar phase POPG bi-layer and is also found to be inducing toroidal-type pore formation.The deduced lipid flip-flop from SFG signals observed from lipids in the C―H and C―D stretching frequency regions also supports MSI-78 induced toroidal-type pore formation.115
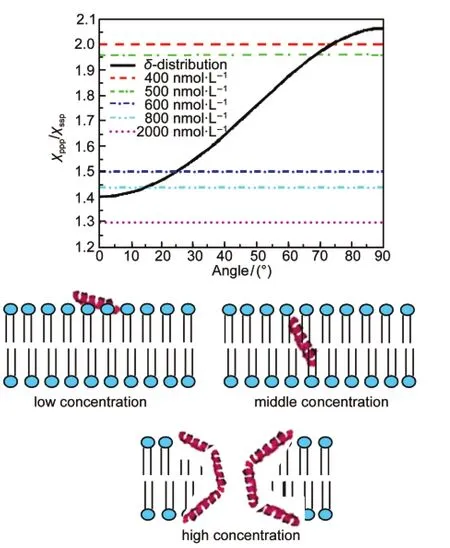
Fig.13 Relation between the measured χppp/χsspratio from polarized SFG spectra and the orientation angle of MSI-78 molecules in a DPPG/d-DPPG bilayer(top); Schematics showing MSI-78 in the lipid layer with different concentrations in the subphase(bottom)Reproduced with permission from reference115.Copyright 2011Am.Chem.Soc.
3.1.3 Melittin
In the above studies on magainin 2 and MSI-78,for the most cases the peptide molecules adopt more or less a single orientation in the lipid bilayer,either nearly parallel,tilt,or nearly perpendicular to the lipid bilayer.However,it was found that under certain conditions the orientation distribution may contain multiple distinct orientations,e.g.,MSI-78 in a DPPG bilayer when the peptide concentration is high.In order to quantify such“multiple-orientation”distributions,we need to measure more independent orientation parameters in the experiments. We therefore combined SFG and ATR-FTIR experiments to measure multiple-orientation distributions of melittin molecules,which adopt an α-helical structure in the lipid bilayer.
Melittin is naturally found in bee venom toxin.It is composed of 26 amino acid residues and is highly positively-charged in physiological conditions.117It can disrupt both bacterial and mammalian cell membranes.Since SFG and ATR-FTIR measure different orientation parameters,118using combined SFG and ATR-FTIR studies,we successfully deduced a more complicated orientation distribution of melittin, which we believe provides a better model for melittin/bilayer interactions,with a DPPG bilayer as an example.98We found that widely used single δ-function and single Gaussian distributions cannot satisfy the observed SFG amide I intensity ratios in spectra collected using different polarization combinations, and thus cannot be used to describe melittin orientation distribution.A maximum entropy trial function has been used to fit the experimental results obtained from the SFG intensity ratio measurement,the absolute SFG intensity measurement,and the polarized ATR-FTIR measurement.98Interestingly,the distribution so determined has two maxima with narrow distribution widths,supporting the idea that melittin helices exist in two main populations in the lipid bilayer at the solution concentration studied(Fig.14).98About three quarters of melittin molecules orient parallel to the bilayer surface with a slight tilt (more or less lie down),while the rest orient more or less parallel to the surface normal(more or less stand up).98Such details have not been revealed before,and cannot be deduced by SFG or ATR-FTIR alone.This work clearly demonstrates the advantages of combining linear and nonlinear vibrational spectroscopies to deduce the melittin orientation distribution.Similarly, this approach can be used to deduce orientations of multiple α-helices in a membrane protein.Such information can be used to determine the membrane protein structure.
3.1.4 Cytochrome b5
The above discussions have been focused on the studies of α-helical peptides,validating the data analysis methodology to study α-helical structures using SFG and combined SFG/ ATR-FTIR methods.We have also applied SFG to examine α-helical structures in proteins to obtain orientation information of proteins in interfacial environments.
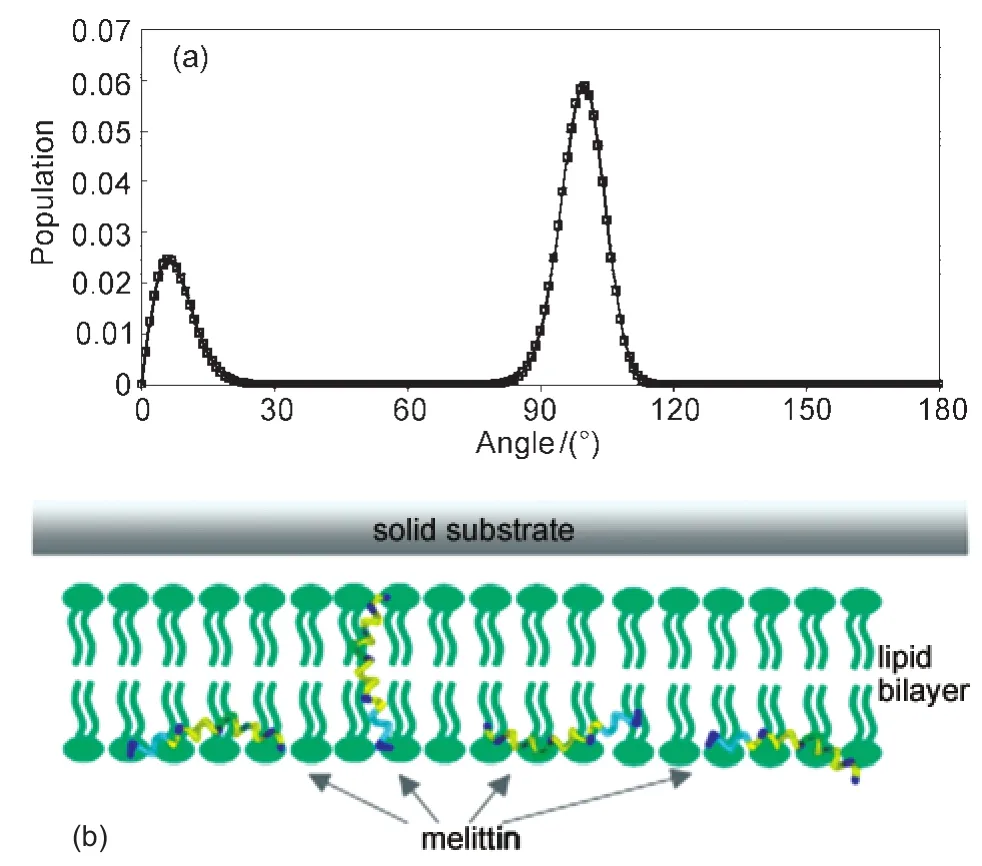
Fig.14 (a)Orientation distribution function of melittin derived based on the maximum entropy theory and(b)schematic of melittinʹs two orientations in the lipid bilayerReproduced with permission from reference98.Copyright 2007Am.Chem.Soc.
To demonstrate the effectiveness of SFG on membrane protein studies,cytochrome b5(Cyt-b5)and its inactive mutants were used as model systems.119Cyt-b5is a 16 kDa tail-anchored membrane protein whose interaction with cytochrome P450 is crucial in drug metabolism.120-122Cyt-b5comprises of three distinct domains with vastly different dynamics:a heme-containing soluble domain,a membrane-spanning anchor,and a linker region connecting the former two.It has been shown that the membrane anchor domain is α-helical in cell membranes.The soluble domain also contains helical structures,but these helical structures more or less point to different directions,and would not generate noticeable SFG signals.We proved that SFG amide I signals from α-helical structures in Cyt-b5are dominated by the contributions from the membrane anchor domain.119
In order to understand the membrane insertion mechanisms of Cyt-b5,we investigated the wild-type Cyt-b5and a Cyt-b5mutant(m-Cyt-b5),in which eight amino acids in the linker region were deleted.119It was interested to observe using SFG that the wild type Cyt-b5can directly insert into a DMPC bilayer(Fig.15),119while the mutant Cyt-b5could not,tilting at about 75°from the membrane normal(Fig.16).119To further investigate the influence of the linker length on the membrane insertion property of Cyt-b5,several Cyt-b5mutants that differ in their linker length were used.SFG results on different mutants in a DMPC bilayer at 25°C inferred that the length of the linker region can indeed influence its membrane insertion:as the length of the linker region increased,the tilt angle of the helical membrane anchor decreased,indicative of membrane insertion(Fig.16).When the temperature is increased,the mutant Cyt-b5which has eight amino acids deleted in the linker region can start to insert into the DMPC bilayer.119We believe that this is due to the fact that there is a kinetic barrier which prevents m-Cyt-b5from penetrating into the hydrophobic region of the bilayer at 25°C.In order to overcome this barrier for insertion to occur,a range of molecular motions is required that permits reorientation,permeation,and translocation of the m-Cyt-b5helical anchor into the membrane.Clearly,the presence of the linker region can make the protein more flexible and increase the mobility of the protein to overcome this kinetic barrier.In fact,it is the length of the linker that influences the membrane insertion property of Cyt-b5as shown in our experimental data as well as the functional properties of the mutant proteins. Therefore,the synergy between the various domains holds the key in the spontaneous membrane insertion of Cyt-b5.
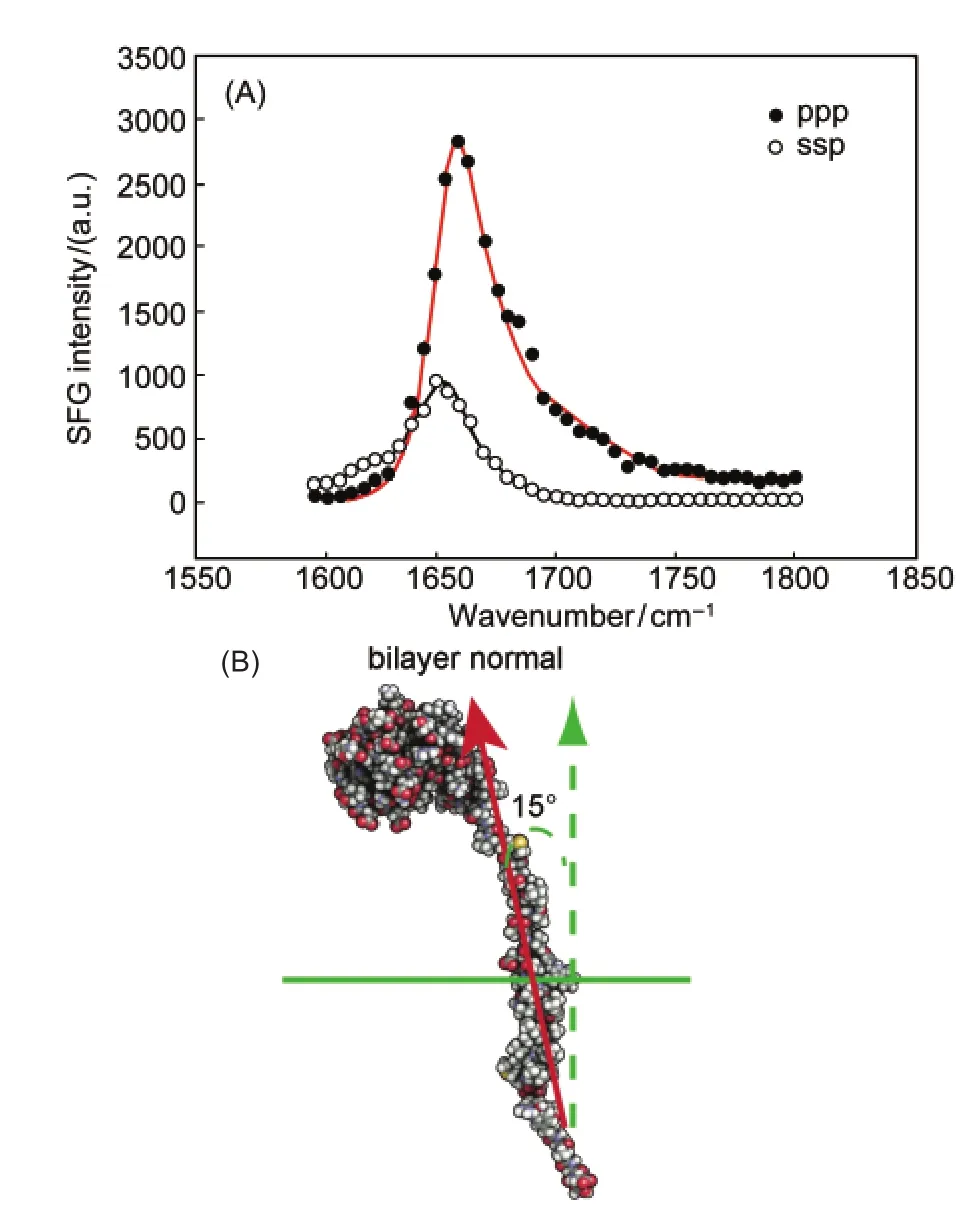
Fig.15 (A)The ssp and ppp polarized SFG amide I signals of Cyt-b5in a dDMPC/dDMPC lipid bilayer at 25°C.From the experimentally measured ppp/ssp ratio,it is possible to calculate the tilt angle of an alpha helix from an SFG experiment; (B)a proposed model of Cyt-b5describing its orientation andtopology in lipid bilayersReproduced with permission from reference119.Copyright 2010Am.Chem.Soc.
3.1.5 G-proteins
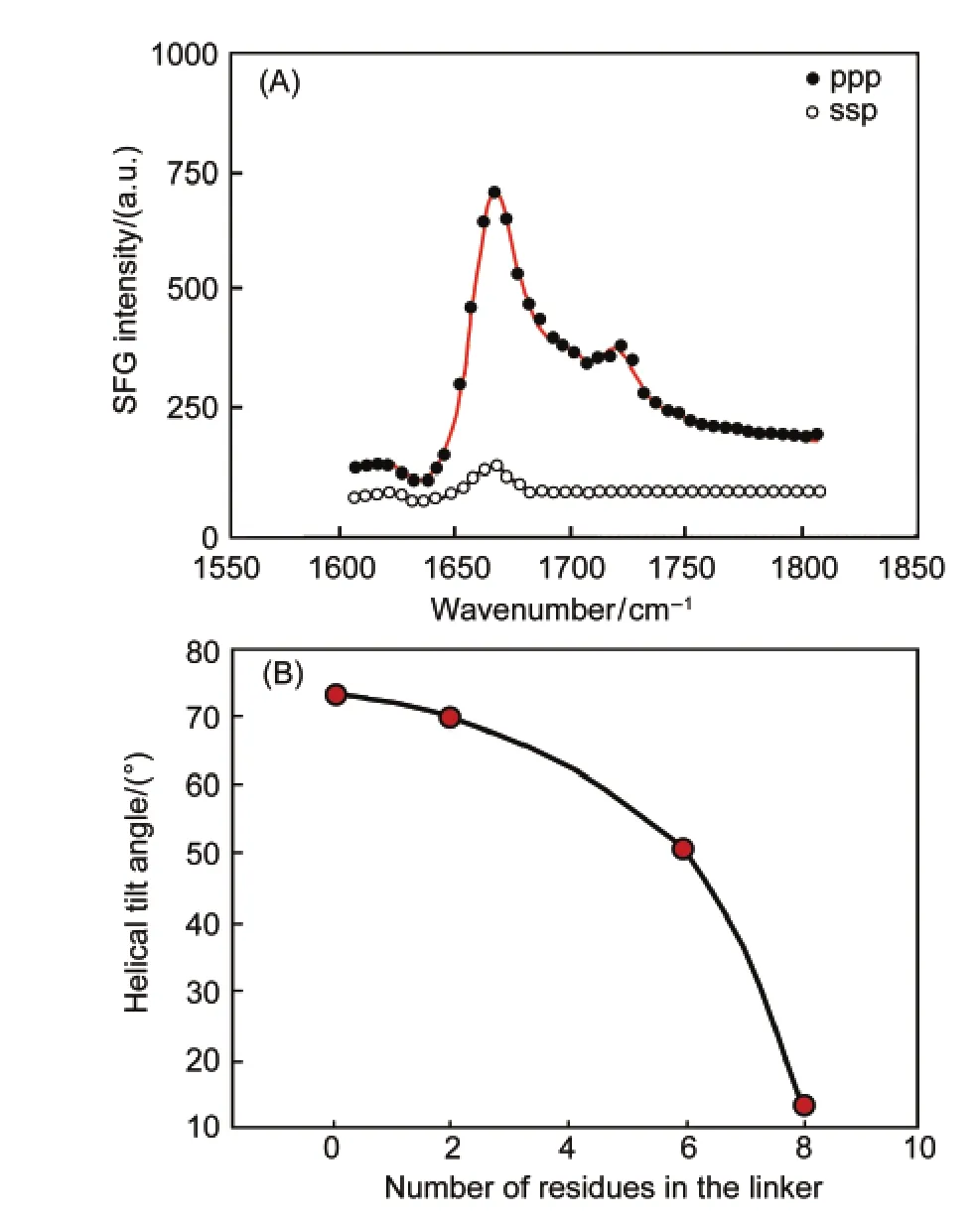
Fig.16 (A)The ssp and ppp polarized SFG amide I signals of a mutant-Cyt-b5in a dDMPC/dDMPC lipid bilayer at 25°C; (B)the dependence of the experimentally measured tilt angle of the transmembrane helix on the number of residues in the linker region of the proteinReproduced with permission from reference119.Copyright 2010Am.Chem.Soc.
Heterotrimeric guanine nucleotide-binding proteins(G proteins)are involved in numerous signal transduction pathways,123,124and relay the extracellular signals sensed by G protein-coupled receptors(GPCRs)to down stream effectors.Heterotrimeric G proteins are comprised of three subunits Gα,Gβ, and Gγ,with Gβ and Gγ forming a tightly associated dimer. SFG has been applied to investigate the membrane orientation of the Gβγ subunit,using lipid bilayers as models for cell membranes.125This was the first time that SFG was used to deduce the orientation of a peripheral membrane protein in the membrane environment in situ.Fig.17125shows the SFG amide I spectra of POPC:POPC bilayer associated Gβ1γ2collected with ssp and ppp polarization combinations.The signals are dominated by the contributions from the α-helical domains in the protein,and the orientation of the helical domain of Gβ1γ2can therefore be determined according to the signal strength ratio of the ppp and ssp amide I signals.Since the protein structure is quite rigid,the orientation of the protein can therefore be known,as displayed in Fig.17.
It was found that the lipid bilayer composition affects the orientation of the associated Gβ1γ2molecules.SFG spectra of the Gβ1γ2adsorbed in bilayers with pure POPC,POPC:POPG(1:1, w/w),and pure POPG as the outer leaflets respectively(while with inner POPC leaflets)were collected and compared.Similar spectral features were observed from Gβ1γ2regardless of the lipid composition,indicating that the α-helices remain the dominant source of signal.The ppp and ssp spectral intensity ratio changed significantly when the lipid composition was changed.Such changes indicate that the lipid composition can modulate the overall Gβ1γ2molecular orientation.125
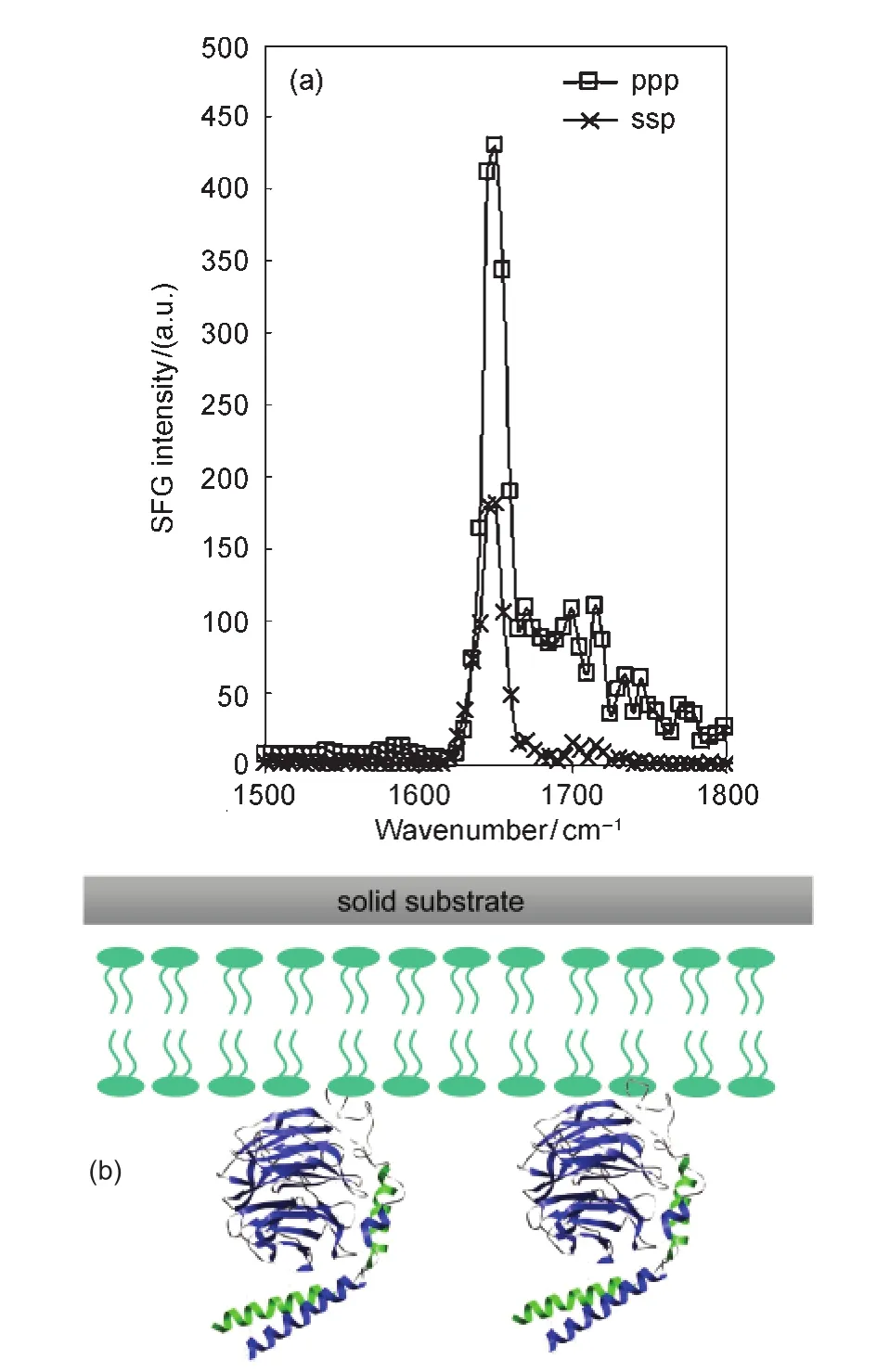
Fig.17 (a)SFG amide I spectra of the interfacial Gβ1γ2adsorbed onto a POPG/POPG bilayer;(b)schematic of Gβ1γ2orientationReproduced with permission from reference125.Copyright 2007Am.Chem.Soc.
SFG was then applied to study the formation of the complex between G protein-coupled receptor(GPCR)kinase 2(GRK2) and Gβ1γ2subunits at a lipid bilayer.126GRK2 is a kinase enzyme responsible for regulating cell signaling.Because of its apparent ability to simultaneously interact with activated heterotrimeric G proteins,G protein-coupled receptors(GPCRs), and the membrane,GRK2 may be involved in the assembly and organization of signaling complexes at GPCRs.Although the atomic structure of GRK2 in complex with both Gαqand Gβγ is known,its orientation while engaged at the cell membrane is less clear because many of the known membrane-binding determinants in this structure are disordered.GRK2 is also an important therapeutic heart disease target.We have developed a computer program that greatly simplifies the analysis of SFG data for any arbitrary protein structure,and that determines the most likely orientations of the protein at a membrane surface.This program can read any arbitrary PDB structure and calculate the polarized SFG signal as a function of net helix orientation.SFG experiments were carried out to observe the Gβγ-GRK complex formation in situ at the lipid bilayer surface.The developed data analysis method was then applied to determine the membrane orientation of the complex(Fig.18).126It was found that the likely membrane orientation of the GRK2-Gβγ complex differs from that predicted from the known protein crystal structure.Instead,a more likely picture holds the predicted receptor docking site of GRK2 in an orientation that would more optimally interact with GPCRs.It was also found that Gβγ appears to change its orientation after binding to GRK2.This research demonstrates that SFG can be used to observe the protein complex formation in situ in real time, and can be used to deduce membrane orientation of large protein complexes.The developed methodology is widely applicable for the study of other membrane proteins in situ.
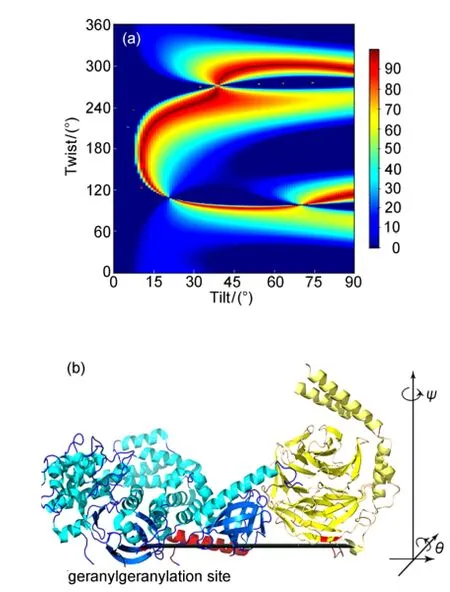
Fig.18 (a)Possible orientation of the Gβγ-GRK2 complex determined by polarized SFG measurements and the use of the computer software;(b)complex at the(0,0)orientationReproduced from reference126.
3.1.6 Fibrinogen
In addition to the membrane associated peptides and proteins discussed above,SFG has also been applied to examine peptides and/or proteins physically adsorbed or chemically immobilized at polymer/solution interfaces.96,127-131Here we discussed the SFG study on fibrinogen as an example.In addition to fibrinogen,SFG has been applied to study albumin,FXII, and chemically immobilized cecropin.
Fibrinogen(~340 kD)is a large blood protein involved in thrombosis.127,128Fig.19127shows the native structure of a trinodular fibrinogen,which has three hydrophobic domains connected by α-helicalcoiled-coils.The conformation ofsurface-bound fibrinogen has been shown to play an important role in thrombus formation.For example,after adsorption to a surface,platelet binding sites may be shielded,reducing the ability to form a thrombus.Therefore it is important to deduce conformation of fibrinogen at interfaces to understand surface biocompatibility and blood coagulation mechanisms.
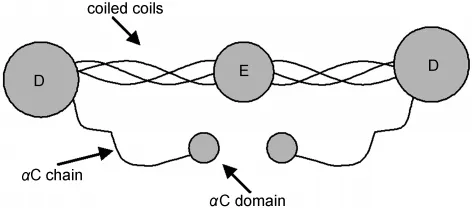
Fig.19 Schematic of fibrinogen native structureReproduced with permission from reference127.Copyright 2005Am.Chem.Soc.
A combined SFG and ATR-FTIR study has been performed to examine fibrinogen adsorption on PS.Fig.19 shows that the native fibrinogen structure has a more or less inversion symmetry,therefore SFG signal should be weak(or even undetectable)if fibrinogen adopts the native structure after the adsorption.Our research showed that very strong SFG signal can be detected from fibrinogen at the PS/solution interface.128We therefore believe that fibrinogen molecules adsorbed on PS adopt bent conformations.The combined polarized SFG and ATR-FTIR studies indicated that the α-helical coiled-coils of fibrinogen exhibit a broad orientation distribution.This broad distribution is caused by adsorbed fibrinogen molecules adopting varied conformations,e.g.,with different angles between two coiled-coils,or different orientations.The PS surface is more complicated than the lipid bilayer environment,therefore induces more complicated interactions with fibrinogen,leading to a broad orientation distribution of fibrinogen coiled coils after adsorption.
3.2 Studies on 310helical structure-alamethicin
In addition to the α-helical structures discussed above,we also studied 310helices using SFG.The data analysis method to determine the orientation of a 310helix at the interface using SFG has been developed,with the same approach used for α-helix orientation determination.132SFG has been applied to characterize interactions between alamethicin(a model for larger channel proteins)and different lipid bilayers in the absence of membrane potential.It was found that alamethicin adopts a mixed α-helical and 310-helical structure in fluid-phase lipid bilayers.The helix(mainly α-helix)at the N-terminus tilts at about 63°versus the surface normal in a fluid-phase 1,2-Dimyristoyl-1D54-sn-Glycero-3-Phosphocholine-1,1,2,2-D4-N,N,N-trimethyl-D9(d-DMPC)/1,2-Dimyristoyl-sn-Glycero-3-Phosphocholine(DMPC)bilayer.The 310helix at the C-terminus (beyond the Pro14 residue)tilts at about 43°versus the surface normal.These results were confirmed by polarized ATR-FTIR studies.This was the first time to apply SFG to study 310helix experimentally.When interacting with a gel-phase lipid bilayer,alamethicin lies down on the gel-phase bilayer surface and/ or aggregates,which does not have significant insertion into the lipid bilayer(Fig.20).132
3.3 Studies on β-sheet structure-tachyplesin I
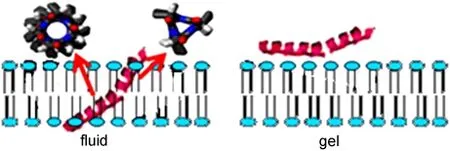
Fig.20 Schematic showing that alamethicin interacts with the fluid phase and gel phase lipid bilayers differentlyReproduced with permission from reference132.Copyright 2010Am.Chem.Soc.
Besides the helical structures,we also applied SFG to investigate orientations of β-sheet structures at interfaces,using tachyplesin I as an example.95,99Different from helical structures,which only require one tilt angle to determine the orientation,it is necessary to deduce both the tilt and the twist angles to quantify the orientation for a β-sheet structure.Therefore, only one SFG intensity ratio obtained from the ppp and ssp spectra cannot lead to enough measured parameters for β-sheet orientation determination.In our studies,we combined SFG and ATR-FTIR to determine orientation of tachyplesin I at interfaces.Tachyplesin I is known to have a β-sheet structure in aqueous environments and at the solution/membrane interface. The β-sheet structure of tachyplesin I is quite robust because this structure is held together by two disulfide bonds.
For a β-sheet structure which adopts a D2symmetry,the relations between the second order nonlinear susceptibility components and the hyperpolarizability components have been deduced.95Different from the helical structures,the SFG measurements in ppp and ssp polarization combination for theD2symmetry point group are not independent to each other;their intensity ratio stays constant(about 2.0),regardless of the β-sheet orientation.Therefore the signal strength ratio measured in the ssp and ppp polarization combinations cannot be used for orientation determination here.To study β-sheet orientation,we used ssp spectra and chiral spectra,e.g.,spp spectra (Fig.21).95,99
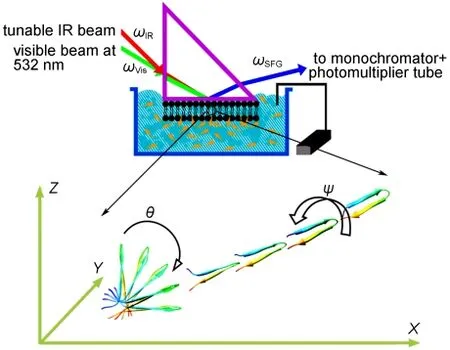
Fig.21 Schematic showing that SFG can be used to study orientation of β-sheet structure at interfacesReproduced with permission from reference95.Copyright 2010Am.Chem.Soc.
Chiral SFG amide I signals were successfully collected from tachyplesin I at interfaces.95,99We believe that these chiral signals are mainly contributed by electric dipole contributions, which can dominate the chiroptical responses of uniaxial systems.We confirmed that chiral SFG signals were contributed by tachyplesin I molecules at interfaces,not from the molecules in the solution bulk.We also showed that the chiral SFG signals were contributed by the β-sheet structures.After using dithiothreitol(DTT)to cleave the di-sulfide bonds in tachyplesin I to destroy the β-sheet structure,no chiral SFG signal can be detected anymore.With SFG ssp and chrial spectra,as well as ATR-FTIR polarized spectra,we quantitatively determined tacyplesin I orientation at the PS/peptide solution interface,as well as in a single supported DPPG bilayer.The tilt and twist angles for tachyplesin I at the PS/tachyplesin I solution interface and in the lipid bilayer are different,due to varied interactions in the two cases.
3.4 Polarization mapping
Only reliable SFG spectral fitting results can lead to correct interpretation of surface/interfacial structures.SFG spectra collected from polymer interfaces and biological interfaces can be very complicated.We developed a polarization mapping method to reliably fit the complex SFG spectra.133In the SFG polarization mapping experiments,we set the polarization of the IR beam at the p polarization direction and the visible beam at 45° from the s polarization direction.The SFG spectra are collected at various polarization angles of a polarizer in front of the detector,thereby detecting SFG signals with different polarizations.133When we set the last polarizer at the s position or p position,the spectra are the same as the SFG spectra collected using the ssp or ppp polarization combinations,except that the intensity decreases to half.If we vary the polarization angle from 0°to 180°,the SFG spectra are interference results of the ssp and ppp spectra.133Since the intensity ratio and phase between ssp and ppp components of the second order nonlinear susceptibility for different vibrational modes are usually different,SFG spectra with very diverse spectral features can be collected by varying the polarization angle.Two-dimensional(2D)SFG spectra can then be constructed by plotting the different SFG spectra as a function of polarization angle(e.g.Fig.22133).If fitting parameters can fit such a two-dimensional SFG spectra reasonably well,such a fitting is usually correct.The polarization mapping method can be used to validate and improve SFG spectral analysis.We can interpret the previous discussion in the following simple way:because SFG is a coherent technique,different vibrational modes detected can interfere with each other.Such interference depends on the structure(e.g.orientation)of different functional groups generating different vibrational modes.By continually changing polarization angles while collecting SFG spectra,we can get different interference patterns of different modes.If we can fit such different complicated interference results,the fitting is usually correct.The polarization mapping method is being applied to test the fitting results of many complicated polymer and biological surfaces and interfaces.
Fig.22 (a)2D polarization mapping spectra collected from ubiquitin at the d-PS/ubiquitin solution interface;(b)fitted resultThe fitting matches the experimental data quite well.Reproduced with permission from reference133.Copyright 2004Am.Chem.Soc.
4 Future
Since the first SFG paper was published in 1987,134,135SFG has been more and more widely used to investigate many different surfaces and interfaces.It has been developed into a powerful technique to elucidate molecular structures of surfaces and interfaces especially buried interfaces in situ in real time.Currently I believe that more than fifty research groups worldwide are carrying out SFG studies.This number will grow substantially in the future,because of the recent demonstrations of the power and uniqueness of the applications of the SFG technique.At the same time,due to the rapid development in laser techniques,detection systems,and optical components,SFG equipment will become more and more user friendly.Very likely in the near future,it will be viewed as a“routine”analytical tool rather than a complicated instrument.
In my group at the University of Michigan,we will continue to focus on the SFG studies on polymer interfaces and biological interfaces.Especially,we will continue to study correlations between buried polymer structures and adhesion,developing SFG into a powerful and nondestructive analytical tool to study industrially important interfaces,such as interfaces involved in applications of polymer adhesives,packaging for microelectronics,and interconnects.Simultaneously,we will further study surface structures of new polymer materials in water to understand their improved biocompatibility or marine antifouling characters.On the other hand,we will progress our studies on membrane peptides and membrane proteins.We will also investigate structure-function relations of chemically immobilized peptides and proteins on surfaces for biosensing as well as other applications.
We recently demonstrated that in addition to collect SFG spectra,we can also collect coherent anti-Stokes Raman scattering(CARS)spectra using the SFG spectrometers in our lab.136To do so,we used the output from the OPG/OPA system as the Stokes beam,and the 532 nm visible beam as the pump/ probe beam.An additional delay line was used to provide temporal overlap of the input beams on the sample when shifting from SFG to CARS spectral collection.Flip mirrors were used to change between SFG and CARS spectral collections.We can collect SFG and CARS vibrational spectra from the same sample to probe its surface and bulk structures without moving the sample.In the future,surface and bulk structures of many different materials will be investigated in the exact same environment.
5 Conclusions
SFG has been developed into a powerful technique to probe molecular structures of polymer and biological interfaces.It can measure vibrational spectra of surfaces and buried interfaces in situ in real time with a submonolayer surface specificity. SFG results indicated that different polymer surfaces can exhibit varied surface restructuring behaviors in water.Therefore in order to understand polymer surface structures in water,polymer surfaces need to be studied in situ.We cannot predict polymer surface structures in water from the surface structures probed in air or in vacuum.Molecular interactions between polymers and a variety of silane molecules serving as model adhesive promoters can be probed in situ at the buried interfaces using SFG.This shows that SFG can be used as a tool to study polymer adhesion at buried interfaces.Molecular structures of buried polymer/polymer and polymer/metal interfaces have also been investigated using SFG.Molecular interactions at these buried interfaces can be determined from SFG results.
It has been demonstrated that SFG is a unique technique to determine membrane orientation of various α-helical peptides such as magainin 2,MSI-78,and melittin in situ.SFG has also been applied to study orientations of larger membrane proteins with α-helical structures such as cytochrome b5,Gβγ,and Gβγ-GRK2 complex.The protein complex formation at model cell membrane surface has been observed in situ in real time using SFG.In addition to the α-helical peptides,methods to deduce membrane orientations of the 310helical structure and the β-sheet structure using SFG have also been developed,and applied to examine membrane orientation of alamethicin and tachyplesin I.Besides membrane peptides and proteins,SFG has also been used to investigate physically adsorbed and chemically immobilized peptides and proteins at polymer/solution interfaces.Molecular level understanding on peptide and protein structures at interfaces using SFG will provide a foundation to understand molecular mechanisms of biocompatibility,marine biofouling,membrane protein functions,biosensing, and antimicrobial peptide activity and selectivity.
Acknowledgments: I appreciate the support from the University of Michigan and want to thank my postdoctoral researchers,graduate students,and collaborators for their contributions to the research.I also want to thank Mr.ZHANG Chi and Dr. BOUGHTONAndrew for designing the cover pictures.
(1) Woodruff,D.;Delchar,T.Modern Techniques of Surface Science;Cambridge University Press:Cambridge,1986.
(2) Somorjai,G.A.Introduction to Surface Chemistry and Catalysis;Wiley:New York,1994.
(3) Shen,Y.R.The Principles of Nonlinear Optics;Wiley:New York,1984.
(4) Chen,Z.;Shen,Y.R.;Somorjai,G.A.Ann.Rev.Phys.Chem. 2002,53,437.
(5) Chen,C.Y.;Liu,W.T.;Pagliusi,P.;Shen,Y.R. Macromolecules 2009,42,2122.
(6) Rao,Y.;Comstock,M.;Eisenthal,K.B.J.Phys.Chem.B 2006, 110,1727.
(7) Moore,F.G.;Richmond,G.L.Accounts Chem.Res.2008,41, 739.
(8) Chen,Z.;Gracias,D.H.;Somorjai,G.A.Appl.Phys.B 1999, 68,549.
(9) Gracias,D.H.;Chen,Z.;Shen,Y.R.;Somorjai,G.A.Accounts Chem.Res.1999,320,930.
(10) Shultz,M.J.;Schnitzer,C.;Simonelli,D.;Baldelli,S.Int.Rev. Phys.Chem.2000,19,123.
(11) Gopalakrishnan,D.;Liu,D.F.;Allen,H.C.;Kuo,M.;Shultz, M.J.Chem.Rev.2006,106,1155.
(12) Kim,J.;Cremer,P.S.J.Am.Chem.Soc.2000,122,12371.
(13) Briggman,K.A.;Stephenson,J.C.;Wallace,W.E.;Richter,L. J.J.Phys.Chem.B 2001,105,2785.
(14) Gautam,K.S.;Schwab,A.D.;Dhinojwala,A.;Zhang,D.; Dougai,S.M.;Yeganeh,M.S.Phys.Rev.Lett.2000,85,3854.
(15) Baldelli,S.J.Phys.Chem.B 2003,107,6148.
(16) Xu,M.;Spinney,R.;Allen,H.C.J.Phys.Chem.B 2009,113, 4102.
(17) Liu,J.;Conboy,J.C.J.Am.Chem.Soc.2004,126,8894.
(18) Ye,H.K.;Gu,Z.Y.;Gracias,D.H.Langmuir 2006,22,1863.
(19) Jayathilake,H.D.;Zhu,M.H.;Rosenblatt,C.;Bordenyuk,A. N.;Weeraman,C.;Benderskii,A.V.J.Chem.Phys.2006,125, 064706.
(20) Stokes,G.Y.;Buchbinder,A.M.;Gibbs-Davis,J.M.;Scheidt, K.M.;Geiger,F.M.J.Phys.Chem.A 2008,112,11688.
(21) Esenturk,O.;Walker,R.A.Chem.Phys.2006,125,174701.
(22) Perry,A.;Neipert,C.;Space,B.;Moore,P.B.Chem.Rev.2006, 106,1234.
(23) Li,Q.F.;Hua,R.;Cheah,I.J.;Chou,K.C.J.Phys.Chem.B 2008,112,694.
(24) Li,Q.F.;Hua,R.;Chou,K.C.J.Phys.Chem.B 2008,112, 2315.
(25) McGall,S.J.;Davies,P.B.;Neivandt,D.J.J.Phys.Chem.A 2005,109,8745.
(26) Chen,P.;Kung,K.Y.;Shen,Y.R.;Somorjai,G.A.Surf.Sci. 2001,494,289.
(27) Chen,Z.Polym.Inter.2006,56,577.
(28) Chen,X.;Chen,Z.Biochim.Biophys.Acta 2006,1758,1257.
(29) Ye,S.;Nguyen,K.;Le Clair,S.V.;Chen,Z.J.Struct.Biol. 2009,168,61.
(30) Le Clair,S.V.;Nguyen,K.;Chen,Z.J.Adhesion 2009,85,484.
(31) Chen,Z.Prog.Polym.Sci 2010,35,1376.
(32) Wang,J.;Woodcock,S.E.;Buck,S.M.;Chen,C.;Chen,Z.J. Am.Chem.Soc.2001,123,9470.
(33) Wang,J.;Chen,C.;Buck,S.M.;Chen,Z.J.Phys.Chem.B 2001,105,12118.
(34) Ratner,B.D.;Castner,D.G.Surface Modification of Polymeric Biomaterials;Plenum Press:New York,1996.
(35) Krishnan,S.;Weinman,C.J.;Ober,C.K.J.Mater.Chem.2008, 18,3405.
(36) Yebra,D.M.;Kiil,S.;Dam-Johansen,K.Prog.Org.Coat. 2004,50,75.
(37) Yoda,R.J.Biomater.Sci.-Polym.Ed.1998,9,561.
(38) Chambers,L.D.;Stokes,K.R.;Walsh,F.C.;Wood,R.J.K. Surf.Coat.Technol.2006,201,3642.
(39) Hron,P.Polym.Inter.2003,52,1531.
(40) Ruckenstein,E.;Gourisankar,S.V.J.Colloid Interface Sci. 1986,109,557.
(41) Yasuda,H.;Charlson,E.J.;Charlson,E.M.;Yasuda,T.; Miyama,M.;Okuno,T.Langmuir 1991,7,2394.
(42) Yasuda,T.;Okuno,T.;Yasuda,H.Langmuir 1994,10,2435.
(43) Hogt,A.H.;Gregonis,D.E.;Andrade,J.D.;Kim,S.W.; Dankert,J.;Feijen,J.J.Colloid Interface Sci.1985,106,289.
(44) Lewis,K.B.;Ratner,B.D.J.Colloid Interface Sci.1993,159, 77.
(45) Zhang,D.;Ward,R.S.;Shen,Y.R.;Somorjai,G.A.J.Phys. Chem.B 1997,101,9060.
(46) Wang,J.;Paszti,Z.;Even,M.A.;Chen,Z.J.Am.Chem.Soc. 2002,124,7016.
(47) Chen,C.Y.;Clarke,M.L.;Wang,J.;Chen,Z.Phys.Chem. Chem.Phys.2005,7,2357.
(48) Clarke,M.L.;Chen,C.;Wang,J.;Chen,Z.Langmuir 2006,22, 8800.
(49) Lu,X.;Clarke,M.L.;Li,D.;Wang,X.;Xue,G.;Chen,Z. J.Phys.Chem.C 2011,115,13759.
(50)Woodcock,S.E.;Chen,C.;Chen,Z.Langmuir 2004,20,1928.
(51) Kristalyn,C.B.;Lu,X.;Weinman,C.J.;Ober,C.K.;Kramer, E.J.;Chen,Z.Langmuir 2010,26,11337.
(52) Chen,C.;Wang,J.;Chen,Z.Langmuir 2004,20,10186.
(53) Ye,S.;Majumdar,P.;Chisholm,B.;Stafslien,S.;Chen,Z. Langmuir 2010,26,16455.
(54) Chen,S.F.;Li,L.Y.;Zhao,C.;Zheng,J.Polymer 2010,51, 5283.
(55) Jiang,S.;Cao,Z.Q.Adv.Mater.2010,22,920.
(56) Zhang,Z.;Finlay,J.A.;Wang,L.F.;Gao,Y.;Callow,J.A.; Callow,M.E.;Jiang,S.Langmuir 2009,25,13516.
(57) Shi,Q.;Ye,S.;Spanninga,S.A.;Su,Y.;Jiang,Z.;Chen,Z.Soft Matter 2009,5,3487.
(58) Chen,C.;Loch,C.L.;Wang,J.;Chen,Z.J.Phys.Chem.B 2003,107,10440.
(59) Chen,C.;Wang,J.;Loch,C.L.;Ahn,D.;Chen,Z.J.Am.Chem. Soc.2004,126,1174.
(60) Loch,C.L.;Ahn,D.;Vázquez,A.V.;Chen,Z.J.Colloid Interface Sci.2007,308,170.
(61) Loch,C.L.;Ahn,D.;Wang,J.;Chen,C.;Chen,Z.Langmuir 2004,20,5467.
(62) Loch,C.L.;Ahn,D.;Chen,C.;Chen,Z.J.Adhesion 2005,81, 319.
(63) Loch,C.L.;Ahn,D.;Chen,Z.J.Phys.Chem.B 2006,110,914.
(64) Vázquez,A.V.;Shephard,N.E.;Steinecker,C.L.;Ahn,D.; Spanninga,S.;Chen,Z.J.Colloid Interface Sci.2009,331,408.
(65) Mine,K.;Nishio,M.;Sumimura,S.Heat Curable Organopolysiloxane Compositions ContainingAdhesion Additives.US Patent 4033924,1977-07-05.
(66) Schulz,J.B.Self-adhering Silicone Compositions and Preparations Thereof.US Patent 4087585,1978-05-02.
(67) Chen,C.;Wang,J.;Even,M.A.;Chen,Z.Macromolecules 2002,35,8093.
(68) Lu,X.;Shephard,N.;Han,J.;Xue,G.;Chen,Z. Macromolecules 2008,41,8770.
(69) Lu,X.;Li,D.;Kristalyn,C.B.;Han,J.;Shephard,N.;Rhodes, S.;Xue,G.;Chen,Z.Macromolecules 2009,42,9052.
(70) Harp,G.P.;Rangwalla,H.;Li,G.;Yeganeh,M.S.;Dhinojwala, A.Macromolecules 2006,39,7464.
(71) Li,G.;Dhinojwala,A.;Yeganeh,M.S.J.Phys.Chem.B 2009, 113,2739.
(72) Yurdumakan,B.;Nanjundiah,K.;Dhinojwala,A.J.Phys. Chem.C 2007,111,960.
(73) Wilson,P.T.;Briggman,K.A.;Wallace,W.E.;Stephenson,J. C.;Richter,L.J.Appl.Phys.Lett.2002,80,3084.
(74) Kweskin,S.J.;Komvopoulos,K.;Somorjai,G.A.Langmuir 2005,21,3647.
(75) Morita,S.;Ye,S.;Li,G.;Osawa,M.Vib.Spectr.2004,35,15.
(76) Ye,S.;Morita,S.;Li,G.;Noda,H.;Tanaka,M.;Uosaki,K.; Osawa,M.Macromolecules 2003,36,5694.
(77) Kweskin,S.J.;Komvopoulos,K.;Somorjai,G.A.Appl.Phys. Lett.2006,88,134105.
(78) Chen,Z.;Ward,R.;Tian,Y.;Malizia,F.;Gracias,D.H.;Shen, Y.R.;Somorjai,G.A.J.Biomed.Mater.Res.2002,62,254.
(79) Mermut,O.;Phillips,D.C.;York,R.L.;McCrea,K.R.;Ward, R.S.;Somorjai,G.A.J.Am.Chem.Soc.2006,128,3598.
(80) Phillips,D.C.;York,R.L.;Mermut,O.;McCrea,K.R.;Ward, R.S.;Somorjai,G.A.J.Phys.Chem.C 2007,111,255.
(81) York,R.L.;Browne,W.K.;Geissler,P.L.;Somorjai,G.A.Isr. J.Chem.2007,47,51.
(82) Weidner,T.;Apte,J.S.;Gamble,L.J.;Castner,D.G. Langmuir 2010,26,3433.
(83) Weidner,T.;Breen,N.F.;Li,K.;Drohny,G.P.;Castner,D.G. Proc.Natl.Acad.Sci.U.S.A.2010,107,13288.
(84)Fu,L.;Ma,G.;Yan,E.C.J.Am.Chem.Soc.2010,132,5405.
(85)Jung,S.Y.;Lim,S.M.;Albertorio,F.;Kim,G.;Gurau,M.C.; Yang,R.D.;Holden,M.A.;Cremer,P.S.J.Am.Chem.Soc. 2003,125,12782.
(86) Chen,X.;Sagle,L.B.;Cremer,P.S.J.Am.Chem.Soc.2007, 129,15104.
(87) Hall,S.A.;Jena,K.C.;Trudeau,T.G.;Hore,D.K.J.Phys. Chem.C 2011,115,11216.
(88) Wang,J.;Buck,S.M.;Chen,Z.Analyst 2003,128,773.
(89)Wang,J.;Buck,S.M.;Even,M.A.;Chen,Z.J.Am.Chem. Soc.2002,124,13302.
(90) Wang,J.;Buck,S.M.;Chen,Z.J.Phys.Chem.B 2002,106, 11666.
(91) Wang,J.;Clarke,M.L.;Zhang,Y.;Chen,X.Langmuir 2003, 19,7862.
(92)Wang,J.;Clarke,M.L.;Chen,X.;Even,M.A.;Johnson,W. C.;Chen,Z.Surf.Sci.2005,587,1.
(93) Chen,X.;Clarke,M.L.;Wang,J.;Chen,Z.Intern.J.Mod. Phys.B 2005,19,691.
(94) Nguyen,K.T.;Le Clair,S.V.;Ye,S.;Chen,Z.J.Phys.Chem. B 2009,113,12169.
(95) Nguyen,K.T.;King,J.T.;Chen,Z.J.Phys.Chem.B 2010, 114,8291.
(96) Wang,J.;Lee,S.H.;Chen,Z.J.Phys.Chem.B 2008,112, 2281.
(97) Lee,S.;Wang,J.;Krimm,S.;Chen,Z.J.Phys.Chem.A 2006, 110,7035.
(98) Chen,X.;Wang,J.;Boughton,A.P.;Kristalyn,C.B.;Chen,Z. J.Am.Chem.Soc.2007,129,1420.
(99) Wang,J.;Chen,X.;Clarke,M.L.;Chen,Z.Proc.Natl.Acad. Sci.U.S.A.2005,102,4978.
(100) Krimm,S.;Bandekar,J.Adv.Protein Chem.1986,38,181.
(101) Barth,A.;Zscherp,C.Q.Rev.Biophys.2002,35,369.
(102) Tamm,L.K.;Tatulian,S.A.Q.Rev.Biophys.1997,30,365.
(103) Wang,J.;Even,M.A.;Chen,X.;Schmaier,A.H.;Waite,J.H.; Chen,Z.J.Am.Chem.Soc.2003,125,9914.
(104) Chen,X.;Wang,J.;Sniadecki,J.J.;Even,M.A.;Chen,Z. Langmuir 2005,21,2662
(105) Lad,M.D.;Birembaut,F.;Clifton,L.A.;Frazier,R.A.; Webster,J.R.P.;Green,R.J.Biophys.J.2007,92,3575.
(106) Ludtke,S.;He,K.;Heller,W.;Harroun,T.;Yang,L.;Huang, H.Biochemistry 1996,35,13723.
(107)Chen,F.Y.;Lee,M.T.;Huang,H.W.Biophys.J.2003,84, 3751.
(108) Ludtke,S.;He,K.;Huang,H.Biochemistry 1995,35,16764.
(109) Imura,Y.;Choda,N.;Matsuzaki,K.Biophys.J.2008,95, 5757.
(110) Boughton,A.P.;Andricioaei,I.;Chen,Z.Langmuir 2010,26, 16031.
(111)Mecke,A.;Lee,D.K.;Ramamoorthy,A.;Orr,B.G.; Banaszak-Holl,M.M.Biophys.J.2005,89,4043.
(112) Gregory,S.M.;Pokorny,A.;Almeida,P.F.F.Biophys.J. 2009,96,116.
(113) Nguyen,K.T.;Le Clair,S.V.;Ye,S.;Chen,Z.J.Phys.Chem. B 2009,113,12358.
(114) Murzyn,K.;Pasenkiewicz-Gierula,M.J.Mol.Model.2003,9, 217.
(115)Yang,P.;Ramamoorthy,A.;Chen,Z.Langmuir 2011,27,7760.
(116) Hallock,K.J.;Lee,D.;Ramamoorthy,A.Biophys.J.2003,84, 3052.
(117) Dempsey,C.E.Biochim.Biophys.Acta 1990,1031,143.
(118) Wang,J.;Paszti,Z.;Clarke,M.L.;Chen,X.;Chen,Z.J.Phys. Chem.B 2007,111,6088.
(119) Nguyen,K.;Soong,T.;Im,S.;Waskell,L.;Ramamoorthy,A.; Chen,Z.J.Am.Chem.Soc.2010,132,15112.
(120) Renthal,R.Cell Mol.Life Sci.2010,67,1077.
(121) Colombo,S.F.;Longhi,R.;Borgese,N.J.Cell Sci.2009,122, 2383.
(122) Dürr,U.H.N.;Ramamoorthy,A.;Waskell,L.Biochim. Biophys.Acta 2007,1768,3235.
(123) Neves,S.R.;Ram,P.T.;Iyengar,R.Science 2002,296,1636.
(124)Cabrera-Vera,T.M.;Vanhauwe,J.;Thomas,T.O.;Medkova, M.;Preininger,A.;Mazzoni,M.R.;Hamm,H.E.Endocr.Rev. 2003,24,765.
(125) Chen,X.;Boughton,A.P.;Tesmer,J.J.G.;Chen,Z.J.Am. Chem.Soc.2007,129,12658.
(126) Boughton,A.P.;Yang,P.;Tesmer,V.M.;Ding,B.;Tesmer,J. J.G.;Chen,Z.Proc.Natl.Acad.Sci.U.S.A.2011,108,E667.
(127) Clarke,M.L.;Wang,J.;Chen,Z.J.Phys.Chem.B 2005,109, 22027.
(128) Wang,J.;Chen,X.;Clarke,M.L.;Chen,Z.J.Phys.Chem.B 2006,110,5017.
(129)Ye,S.;Nguyen,K.T.;Boughton,A.P.;Mello,C.M.;Chen,Z. Langmuir 2010,26,6471.
(130) Han,X.;Soblosky,L.;Slutsky,M.;Mello,C.M.;Chen,Z. Langmuir 2011,27,7042.
(131) Chen,X.;Wang,J.;Paszti,Z.;Wang,F.;Schrauben,J.N.; Tarabara,V.V.;Schmaier,A.H.;Chen,Z.Anal.Bioanal. Chem.2007,388,65.
(132)Ye,S.;Nguyen,K.T.;Chen,Z.J.Phys.Chem.B 2010,114, 3334.
(133) Wang,J.;Clarke,M.L.;Chen,Z.Anal.Chem.2004,76,2159.
(134) Guyotsionnest,P.;Hunt,J.H.;Shen,Y.R.Phys.Rev.Lett. 1987,59,1597.
(135) Hunt,J.H.;Guyot-Sionnest,P.;Shen,Y.R.Chem.Phys.Lett. 1987,133,189.
(136)Zhang,C.;Wang,J.;Khmaladze,A.;Liu,Y.;Ding,B.; Jasensky,J.;Chen,Z.Opt.Lett.2011,36,2272.
November 14,2011;Revised:January 2,2012;Published on Web:January 9,2012.
Molecular Structures of Buried Polymer Interfaces and Biological Interfaces Detected by Sum Frequency Generation Vibrational Spectroscopy
CHEN Zhan*
(Department of Chemistry,University of Michigan,930 North University Avenue,Ann Arbor,MI 48109,USA)
Molecular structures at interfaces determine interfacial properties.In order to optimize the interfacial structures to achieve improved properties of advanced materials,it is important to characterize molecular structures of interfaces in situ in real time.Recently,a nonlinear optical spectroscopic technique, sum frequency generation(SFG)vibrational spectroscopy,has been developed into a powerful and unique tool to elucidate molecular structures of buried interfaces,including liquid/liquid,solid/liquid,and solid/solid interfaces.In this review,applications of SFG to study molecular structures of complex interfaces such as polymer interfaces and biological interfaces have been discussed.Particularly,molecular surface structural changes of various polymers in water,molecular interactions between polymers and silane model adhesion promoters at interfaces,and structures of buried polymer/polymer as well as polymer/metal interfaces have been presented.In addition,interfacial structures of peptides with varied secondary structures and several representative proteins have been introduced.Interfaces play important roles in many disciplines ranging from chemistry,biology,physics,materials science and engineering,to nanotechnology.The development of a unique technique which can probe molecular structures of complex interfaces in situ greatly impacts the research in these disciplines as well as many interdisciplinary studies.
Polymer surface and interface;Biological interface;Peptide;Protein;Adhesion; Secondary structure;Alpha-helix;Beta-sheet
10.3866/PKU.WHXB201201091
O647
∗Corresponding author.Email:zhanc@umich.edu;Fax:+1-734-647-4865.
The project was supported by the National Science Foundation,USA(CHE 1111000),National Institute of Health,USA(GM081655),Army Research Office,USA(W911NF-11-1-0251),Defense Threat ReductionAgency,USA(HDTRA1-11-1-0019),Office of Naval Research,USA (N00014-08-1-1211),and Semiconductor Research Corporation,USA(P10419).
