以主族元素为桥的梯形化合物的光电性质
2012-11-30刘淑娟马廷春许文娟刘湘梅黄艳琴
刘淑娟 马廷春 许文娟 刘湘梅 赵 强 黄艳琴 黄 维
(南京邮电大学信息材料与纳米技术研究院,有机电子与信息显示国家重点实验室培养基地,南京210046)
以主族元素为桥的梯形化合物的光电性质
刘淑娟†马廷春†许文娟 刘湘梅 赵 强*黄艳琴 黄 维
(南京邮电大学信息材料与纳米技术研究院,有机电子与信息显示国家重点实验室培养基地,南京210046)
梯形化合物具有大的平面π共轭结构,不会产生构象扭曲,可以有效增加π共轭长度,因而表现出非常好的光电性质.将主族元素引入到梯形化合物骨架中作为桥接单元不仅可以固定其结构而且由于主族元素和π共轭骨架之间的轨道相互作用,可以实现对这类化合物光电性质的调节.采用密度泛函理论对一系列主族元素桥的梯形化合物的结构和光电性质进行了理论研究,从而可更好地理解和预测这类化合物的性质.研究发现,这类化合物的电子结构可以通过引入主族元素进行调节.由于具有更大的π共轭程度,四主族元素桥化合物的吸收与双主族元素桥化合物相比有明显的红移,而且荧光寿命较短.另外,通过计算离子化势(IPs)、电子亲和能(EAs)和重组能(λ)考察了这类化合物的电子和空穴注入及传输性质.研究发现,四主族元素桥化合物表现出更强的电子和空穴注入能力.
密度泛函理论;电子性质;梯形化合物;主族元素;光物理性质
1 Introduction
Novel π-conjugated molecules with desirable electronic and photophysical properties are very important for the advances of organic electronics.Since the flattened π-conjugated framework can eliminate the conformational disorder and effectively enhance the π-conjugation,ladder-type π-conjugated molecules with fully ring-fused structure have fascinating properties of high fluorescence efficiency,excellent thermal stability,and high carrier mobility.1-5Thus,this class of molecules is promising materials for optoelectronic devices,such as organic light-emitting diodes,field effect transistors,etc.An effective way of successfully achieving ladder-type π-conjugated molecules is to incorporate the main group elements into the ladder skeleton,namely heteroatom-bridged homologues,because the heteroatom-bridges would not only stiffen the skeleton but also contribute to the electronic structure through orbital interaction between heteroatoms and π-conjugated skeleton.5A number of ladder-type heteroatom-bridged stilbenes with various heteroatoms,such as group 13 boron,6-9group 14 silicon,10-15group 15 nitrogen16-19and phosphorous,20-24have already been reported. It has been demonstrated that the electronic structures of these π-conjugated skeletons can be tuned by the incorporated elements.It is noteworthy that the third row elements,such as silicon or phosphorus,can significantly improve the electron-accepting ability due to the σ*-π*orbital interaction.9,24
Due to their excellent optoelectronic properties,theoretical investigations on the heteroatom-bridged stilbenes are very necessary to rationalize experimental results of known compounds and to predict those of unknown ones.It is well-known that density functional theory(DFT)and time-dependent DFT (TDDFT)calculations are the most widely used techniques for the theoretical investigation of organic π-conjugated molecules.By DFT/TDDFT,the ground-and excited-state properties,electronic structures,and spectroscopic properties of the π-conjugation molecules can be described and predicted.25-31Furthermore,DFT calculations have also been successfully utilized to predict and analyze the charge accepting and transporting properties of organic luminophores.32-36
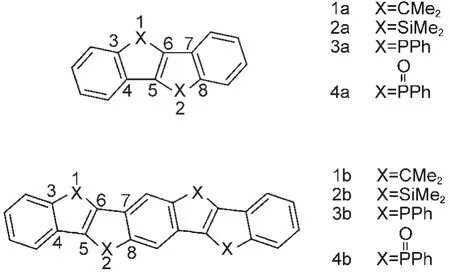
Fig.1 Molecular structures of 1a-4a and 1b-4b
Herein,the structural,electronic,and optical properties of bis-and tetrakis-bridged(C,Si or P-bridged)stilbene derivatives(Fig.1)were investigated by DFT/TDDFT and single excitation configuration interaction to provide theoretical understandings and predictions of these compounds.Tetrakissiliconbridged derivative has been demonstrated to exhibit substantial red shifts in the absorption and emission wavelength maxima without a significant decrease in fluorescence quantum yield because of extended π-conjugation compared with the carbon analogue.12In addition,bis-phosphoryl-bridged stilbenes show intense blue fluorescence at longer wavelengths with higher quantum yields compared with the carbon and silicon analogues.Therefore,investigations on new tetrakis-bridged stilbenes are very interesting and necessary.Their synthetic procedures,however,are often very complicated.Thus theoretical investigations based on quantum chemical calculations will provide efficient way to predict their photophysical and electronic properties.
2 Theoretical and computational methodology
The charge mobility was evaluated by the incoherent hopping model,in which the charge transport is an intermolecular process.The charge transfer rate k can be described as follows according to the Marcus/Hush model.37
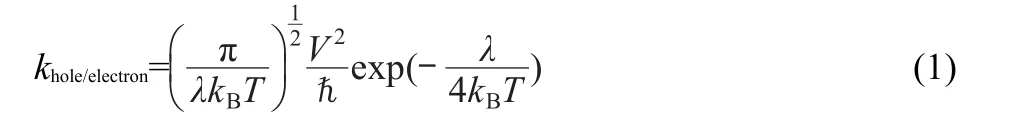
where V is the charge transfer integral between the two species, λ is the reorganization energy,kBis Boltzmann constant,ℏ=h/ 2π,h is Planck constant,and T is the temperature.Thus,the transport process is determined by V and the internal λ.High V and small λ are needed for an efficient charge-transport.38,39It is experimentally determined that V shows a rather narrow range of values and an even more limited range is expected due to the intermolecular charge-transfer processes considered in organic light-emitting diodes(OLEDs)involving direct contacts in amorphous solids.35The reorganization energy for hole/electron transfer can be simply defined as follows:40
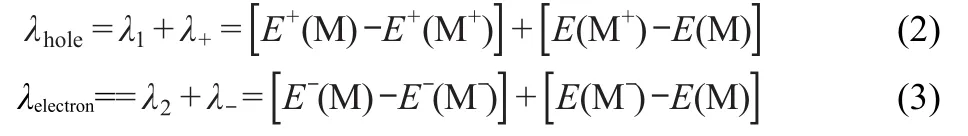
The sum of λ1and λ+is the hole reorganization energy of λhole. Here λ1is the relaxation energy from M+extracting a hole going toward the M optimum geometry on the potential energy surface of M,and λ+is the relaxation energy of a neutral molecule M that captures a hole going toward the M+optimum geometry on the potential energy surface of M+.The calculation method of electron reorganization energy of λelectronis similar to that of λhole.M+and M-denote their optimized structures at corresponding ion states.E,E+,and E-represent the energies of the neutral,cation,and anion species based on their lowest energy geometries,respectively.
All computations were carried out with Gaussian 03 program package41with different parameters for structure optimizations and vibrational analyses.The singlet ground-state geometries were fully optimized by the Beckeʹs three-parameter exchange functional along with the Lee-Yang-Parr correlation functional with the restricted(B3LYP)and the unrestricted formalism(UB3LYP)for neutral and ion state molecules at the standard split valence plus polarization function 6-31G(d)basis set,respectively.42The excited-state geometries were optimized by the ab initio configuration interaction singles method (CIS),which can also be done by using the TDDFT with Gaussian 09 program package now.These optimized stationary points were further characterized by harmonic vibrational frequency analysis to ensure that real local minimum has been found without imaginary vibrational frequency.The electronic absorption and emission spectra were carried out using TDDFT method of B3LYP/6-31G(d)on the basis of the optimized ground and excited state structures,respectively.The polarizable continuum model(PCM)was adopted in the solvent (CH2Cl2and THF).
3 Results and discussion
3.1 Optimized ground-state geometries
The structural parameters of trans-4a have been computed with DFT/B3LYP using different basis sets of 6-31G(d),6-31+ G(d),and 6-311+G(d).Similar structural parameters were obtained as listed in Table S1 from Supporting Information.The deviations of the selected bond lengths and bond angles are no more than 0.005 nm and 0.25°,respectively.It can be seen that the basis set of 6-31G(d)could provide comparable accuracy as the larger basis sets.In addition,The selected important bond lengths as well as related bond angles obtained by calculation of B3LYP/6-31G(d)(Table S1)are quite in agreement with the corresponding experimental results(X-ray crystal diffraction data).24Hence,the method of B3LYP/6-31G(d)was selected for the following calculations.
The stilbene skeletons of these optimized ladder-type molecules exhibit highly coplanar structure due to the heteroatombridges.The front and side views of the optimized ground-state geometries of trans-4a and trans-4b as examples are shown in

Fig.2 Front and side views of the optimized ground-state geometries of trans-4a and trans-4b at the B3LYP/6-31G(d)level
Fig.2.These heteroatom-bridged stilbene homologues are symmetrical and the structural parameters of the cis and trans isomers are very similar with each other.Heteroatoms have effect on the structural parameters of these ladder-type π-conjugated molecules(see Tables S2 and S3).The Si and P related bond lengths become evidently longer(>0.03 nm)than those corresponding bond lengths in 1a and 1b.The bond angles of C―Si―C in 2a,2b as well as C―P―C in 3a,3b and 4a,4b significantly decrease more than 8°compared with C―C1―C in molecule 1.The bond angels of C3―C4―C5 and C4―C5―C6 in 2a-4a,2b-4b,however,significantly increase more than 5°compared with those in 1a,1b.
3.2 Optimized excited-state geometries
To understand the structural differences between the groundand excited-state geometries,the excited-state geometries of 1a-4a are optimized and the selective structural parameters are listed in Table 1.For C-bridged 1a,the bond length of C4―C5 shortens about 0.006 nm,while the bond length of C5―C6 increases 0.007 nm.The bond length of C3―C4 becomes longer with moderate variation of about 0.002 nm.The bond angle of C3―C1―C6 decreases while those of C1―C3―C4 and C3―C4―C5 increase about 1°after they are excited.The changes of other selected bond lengths and bond angles are not significant.For Si-bridged 2a,the bond length of Si―C6 exhibits moderate variation of about 0.002 nm upon excitation.The changes of other bond lengths are very similar to those of 1a.It is noteworthy that the bond angles of C4―C5―C6 exhibit variation of more than 3°after 2a is excited.Upon excitation, the structural parameter changes of 3a and 4a are very similar to those observed in 2a.
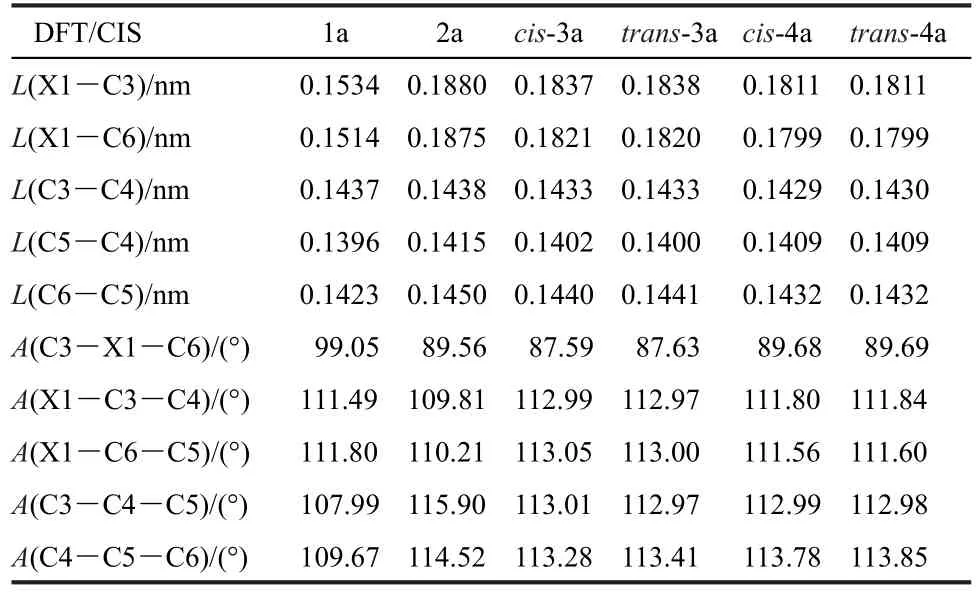
Table 1 Selected bond lengths and bond angles of 1a-4a at the excited state optimized by CIS/6-31G(d)
3.3 Frontier molecular orbitals
Electronic properties of these heteroatom-bridged stilbenes were studied and the orbital energy levels of the highest occupied molecular orbitals(HOMOs)and the lowest unoccupied molecular orbitals(LUMOs)are shown in Fig.3.For both bisand tetrakis-bridged bis(styryl)benzenes,Si-and P-bridged homologues show decreased HOMO and LUMO energies compared with C-bridged homologues,indicating enhanced electron-accepting ability and decreased energy gap(Eg).The cis and trans isomers show similar HOMO and LUMO levels. Due to the similar LUMO levels and lower HOMO levels, P-bridged 3a and 3b exhibit higher hole-accepting ability and Egvalue compared with those of Si-bridged 2a and 2b,respectively.Because of the introduction of strong electron-withdrawing substituent group of O,4a and 4b exhibit dramatically enhanced electron-accepting ability with the significantly decreased energies of HOMO,LUMO,and Egcompared with their analogue.It is obvious that the tetrakis-bridged bis(styryl) benzenes exhibit significantly lower Egwith the decreased LUMO and increased HOMO compared with their bis-bridged analogues due to the extended π-conjugation.
The electronic density contours of the frontier orbitals were plotted by GaussView as shown in Fig.4.For both bis-and tetrakis-bridged homologues,the electronic cloud distributions of HOMOs localize mainly on the stilbene unit.P and O give small contribution to HOMOs for 3a and 4a,3b and 4b,respectively.The electronic cloud distributions of LUMOs are similar to those of HOMOs with the difference of more contributions from P and O for 3a,3b,4a,and 4b,respectively.From this,it can be seen that the introduction of main group atoms,especially P and O,has great influences on the distributions of frontier molecular orbitals and electron transitions.
3.4 Electrical carrier injection and transporting characteristics
The charge injection,transport as well as their balance of optoelectronic materials are crucial for their applications in optoelectronic devices.Therefore,it is important to evaluate the energy barrier for the injection and transport rates of the holes and electrons by investigating the ionization potentials(IPs), electronic affinities(EAs),and reorganization energies(λholeand λelectron)of these compounds together with the hole extraction potential(HEP)and the electron extraction potential (EEP)as listed in Table 2.The IPs and EAs can be either for vertical excitations(v)or adiabatic excitations(a).The lower IP and higher EA,the easier entrance of holes and electrons,respectively.
For bis-bridged derivatives as shown in Table 2,C-bridged 1a exhibits the lowest IP,EA,and reorganization energies,indicating that 1a has higher hole-accepting ability and chargetransport rate compared with Si-or P-bridged 2a-4a.The difference between λholeand λelectronof 1a is 0.07 eV.The electron-accepting ability of 1a,however,is worse due to its low EA, which is in complete accordance with its high LUMO energy level.By substitution of C with Si or P(2a or 3a),the increased IPs and EAs are observed,indicating the higher electron-accepting ability.IPs,especially EAs of cis-and trans-3a are higher than those of 2a.The highest IP and EA are observed for cis-and trans-4a,indicating that the P=O groups significantly facilitate the electron-injection.The λholeis smaller than their respective λelectronfor all compounds,showing that the hole-transfer may be faster than the electron-transfer.Thus,it is obvious that main group atoms Si and P have great influence on the charge injection,transport as well as their balance of optoelectronic materials and the introduction of P can enhance the electron-accepting ability dramatically,which is in accordance with their frontier orbital energy levels.
Compared with bis-bridged derivatives,tetrakis-bridged derivatives exhibit lower IP and higher EA,indicating higher accepting ability of both hole and electron.It is fascinating that λ, especially λelectronof tetrakis-bridged derivatives,decreases significantly,showing the probability of high charge-transport rate.In addition,λholeand λelectronare very close to each other. Thus,tetrakis-bridged derivatives exhibit more promising applications in the optoelectronic field due to their extended π-conjugation.
3.5 Absorption and emission spectra
3.5.1 Absorption spectra

Fig.3 Frontier molecular orbital energies of 1a-4a and 1b-4b

Fig.4 Electron density distributions of the HOMO and LUMO of 1a-4a and 1b-4b
On the basis of the optimized ground-state geometry,the TDDFT method was used to calculate the absorption properties of 1a-4a and 1b-4b in both CH2Cl2and THF solutions.To intuitively understand the absorption properties of these compounds,fitted Gaussian type absorption curves with the calculated absorption data of these compounds in CH2Cl2are shown in Fig.S1.The calculated wavelengths,main transition configurations,and oscillator strengths for the absorption properties are typically listed in Table 3 together with some obtained experimental absorption maxima.12,24From Table 3 and Fig.S1,it can be found that the calculated absorption bands are very similar to experimental data.The absorption behaviors of com-pounds 1a-4a and 1b-4b in THF are very similar to those in CH2Cl2,indicating that solvent has insignificant effect on the absorption properties.
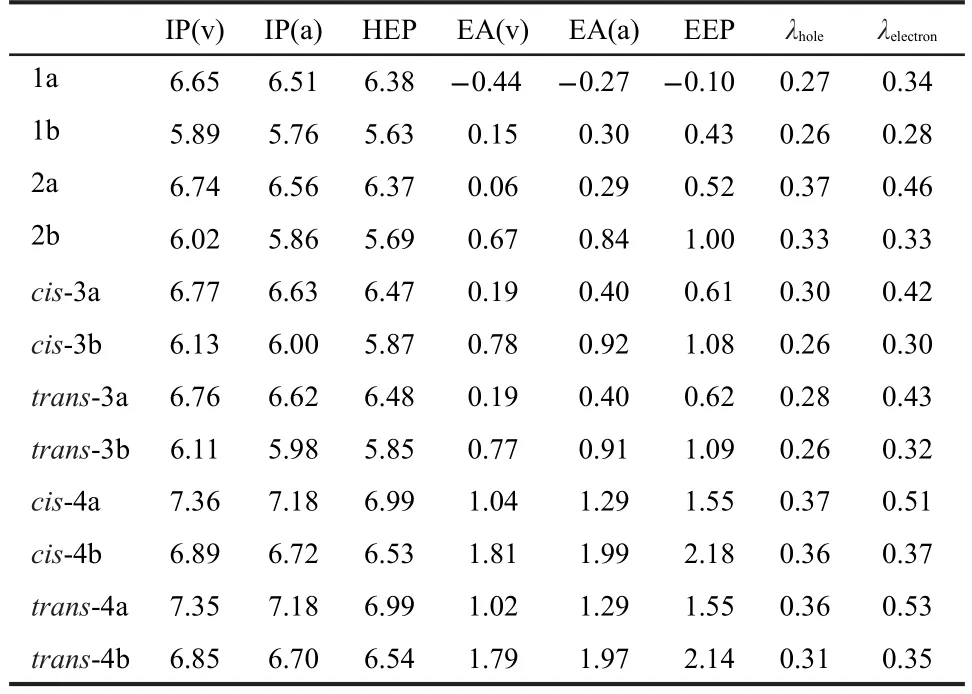
Table 2 Ionization potential(IP),electronic affinity(EA),and reorganization energy(λ)(all in eV)of 1a-4a and 1b-4b
As shown in Table 3,all compounds studied here have the strongest absorption peaks,with large oscillator strengths assigned to S0→S1electronic transition.The lowest singlet excited state corresponds to the excitation from HOMO to LUMO with the largest confguration coefficient for all studied compounds.This is in agreement with the results discussed above that the dominating transition of these compounds will be HOMO→LUMO due to the great difference between HOMO and HOMO-1 as well as LUMO and LUMO+1 energy levels. According to the distributions of HOMOs and LUMOs as shown in Fig.4,the strongest absorption bands are assigned to the π-π*transition.
The C-bridged 1a shows absorption maxima at 332 and 331 nm in CH2Cl2and THF,respectively.The Si-bridged 2a and P-bridged 3a exhibit absorption maxima at 374 and 360 nm,respectively.After chemical modification of P with O(cis-and trans-4a),however,the absorption maxima show evident red-shifts in both CH2Cl2and THF(414-412 nm).From this,it can be seen that the absorption properties are in good agree-ment with the Egof these compounds.It is noteworthy that the obvious red-shifts of the absorption maxima for 2a-4a compared with those of the carbon analogue 1a demonstrate the considerable effect of the main group elements.Compared with bis-bridged analogues 1a-4a,tetrakis-bridged 1b-4b exhibit substantial red shifts in the absorption because of extended π-conjugation.As shown in Fig.S1,the absorption spectra of the cis and trans isomers are almost identical with each other.This indicates that the configuration of the phosphoryl group does not affect the absorption properties.

Table 3 Calculated excited energies,dominant orbital excitations,and oscillator strength of 1a-4a and 1b-4b in THF and CH2Cl2solvents
3.5.2 Emission spectra
The theoretical emission properties for 1a-4a are obtained by TDDFT calculations based on optimized excited-state geometries and the related data are collected in Table 4 together with the experimental emission properties for comparison.The calculated emission spectra for 1a-4a in CH2Cl2solvent were selected to be shown in Fig.S2.We can see that the calculated lowest-energy emissions of 1a-4a exhibit red shifts compared with the experimental emission wavelengths to a certain extent.The calculated lowest-energy emission of 1a in THF(389 nm)is very similar to the corresponding experimental emission wavelength(367 nm).The theoretical calculated lowest-energy emission of 2a in THF shows 45 nm longer than the corresponding experimental emission maximum.The experimentalemission wavelengths of the cis-3a and trans-3a can not be obtained,and the obtained experimental emission wavelength maximum of the mixture of cis and trans isomers(415 nm in CH2Cl2)is closed to their calculated lowest-energy emissions (450 nm for cis-3a and 438 nm for trans-3a in CH2Cl2).The calculated lowest-energy emissions of cis-4a and trans-4a in CH2Cl2exhibit evident deviation of about 60 nm longer than experimental emission wavelength maxima.The discrepancies between the calculated values and experimental data may be attributed to the DFT theory,which generally gives a smaller gap of molecules and thus generates smaller excited energies,especially for large conjugated systems and charge-transfer complexes in excited states.35
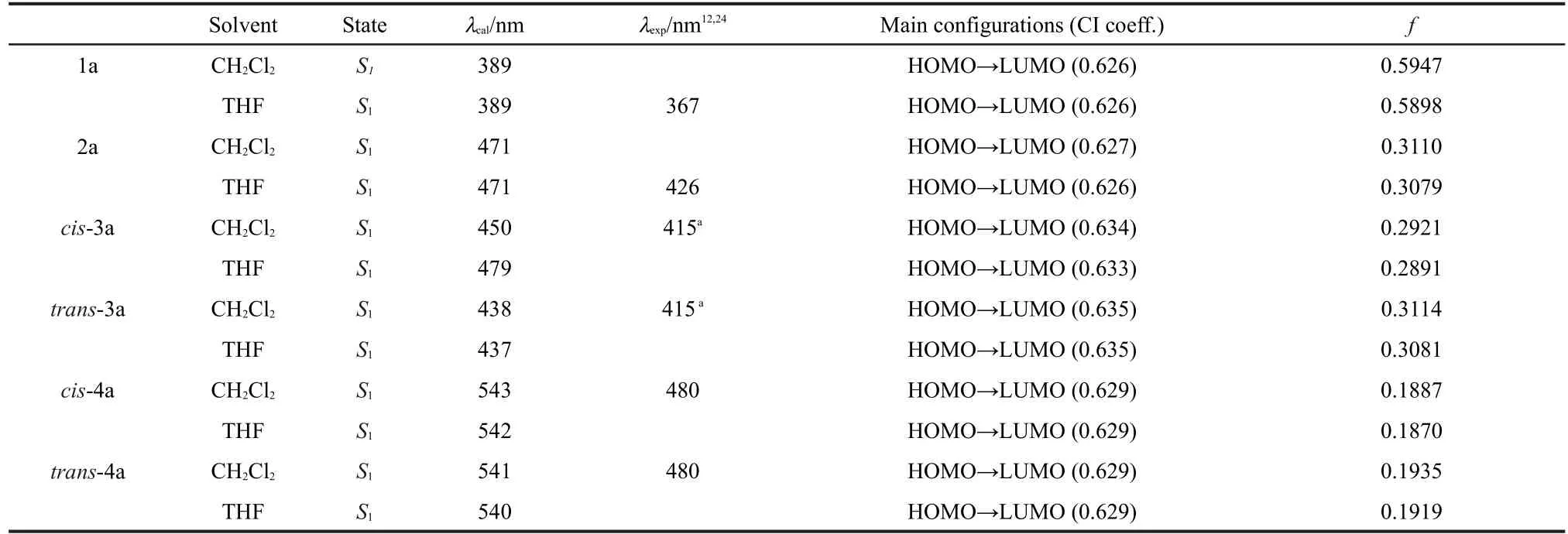
Table 4 Emission properties of heteroatom-bridged stilbenes of 1a-4a in both CH2Cl2and THF solvents
From Table 4 we can see that the emission properties of all compounds studied in CH2Cl2solutions are very similar to those in THF solutions,indicating that the solvent polarity has no evident influences on the emission properties of these compounds.The C-bridged 1a shows deep blue light-emitting features(389 nm)and the emission of Si-and P-bridged compounds exhibit red shifts compared with 1a.Like those observed in the absorption spectra,the emission wavelengths for 1a-4a also exhibit red shifts in the order of 1a<4a<2a<3a.The emission of 1a-4a is all assigned to π→π*character arising from S1state,namely HOMO→LUMO transition.
The radiative lifetimes of these heteroatom-bridged stilbenes were calculated using the Einstein transition probabilities on the basis of excitation energy and oscillator strength according to the formula(in a.u.).35

where c is the velocity of light,EFluis the transition energy,and f is the oscillator strength.The calculated radiative lifetimes together with some experimental data are listed in Table 5.It can be seen that the calculated radiative lifetimes are close to the corresponding experimental lifetimes,12,24suggesting that calculation is reliable.It can ben found that solvents show no obvious effect on the radiative lifetimes,which is consistent withabsorption and emission properties.As compared with the carbon analogue 1a,silicon-and phosphanyl-bridged stilbenes (2a,cis-3a,and trans-3a)exhibit longer radiative lifetimes. Phosphoryl-bridged cis-4a and trans-4a give the longest radiative lifetimes.The trends of radiative lifetimes for the different tetrakis-bridged stilbenes are similar to those of bis-bridged analogues.It is noteworthy that the increase of the π-conjugation using tetrakis-bridges leads to a decrease of excitation energies and a significant increase of oscillator strengths.As a consequence,the radiative lifetimes of tetrakis-bridged stilbenes are shorter than those of their corresponding bis-bridged analogues.

Table 5 Radiative lifetime(τ)of heteroatom-bridged stilbenes of 1a-4a and 1b-4b in both CH2Cl2and THF solvents
4 Conclusions
In conclusion,heteroatom-bridged stilbenes are a class of promising materials with excellent photophysical and electronic properties due to the fully ring-fused polycyclic skeletons. Their structural parameters show obvious changes upon excitation.Electronic properties of these heteroatom-bridged stilbenes were studied and the transition from HOMO to LUMO, assigned to π→π*character,were dominating.It was demonstrated that the electronic structures of these π-conjugated skeletons can be tuned by incorporating main-group elements.The energy barrier for the injection and transport rates of the holes and electrons was evaluated by IPs,EAs,and λ.The main group atoms Si and P have great influence on the charge injection,transport as well as their balance of optoelectronic materials.The introduction of P can enhance the electron-accepting ability dramatically.Compared with bis-bridged analogues,tetrakissilicon-bridged derivatives exhibit substantial red shifts in the absorption because of extended π-conjugation.The increase of the π-conjugation using tetrakis-bridges leads to shorter radiative lifetimes compared with their corresponding bis-bridged analogues.In addition,tetrakissilicon-bridged derivatives exhibit higher accepting ability of both hole and electron.
Supporting Information: The optimized geometries of 1a-4a and 1b-4b at B3LYP/6-31G(d)level,the simulated absorption spectra of 1a-4a and 1b-4b,and the simulated emission spectra of 1a-4a have been included.This information is available free of charge via the internet at http://www.whxb. pku.edu.cn.
(1) Scherf,U.J.Mater.Chem.1999,9,1853.doi:10.1039/ a900447e
(2)Watson,M.D.;Fechtenkötter,A.;Müllen,K.Chem.Rev.2001, 101,1267.doi:10.1021/cr990322p
(3)Bendikov,M.;Wudl,F.Chem.Rev.2004,104,4891.doi: 10.1021/cr030666m
(4)Anthony,J.E.Chem.Rev.2006,106,5028.doi:10.1021/ cr050966z
(5) Fukazawa,A.;Yamaguchi,S.Chem.Asian J.2009,4,1386. doi:10.1002/asia.v4:9
(6) Zhao,Q.;Zhang,H.;Wakamiya,A.;Yamaguchi,S.Synthesis 2009,1,127.
(7)Li,D.;Zhang,H.;Wang,C.;Huang,S.;Guo,J.;Wang,Y. J.Mater.Chem.2012,22,4319.doi:10.1039/c1jm14606h
(8)Agou,T.;Kobayashi,J.;Kawashima,T.Chem.Eur.J.2007,13, 8051.doi:10.1002/(ISSN)1521-3765
(9)Fukazawa,A.;Yamada,H.;Yamaguchi,S.Angew.Chem.Int. Edit.2008,47,5582.doi:10.1002/anie.v47:30
(10)Matsuda,T.;Kadowaki,S.;Goya,T.;Murakami,M.Org.Lett. 2007,9,133.doi:10.1021/ol062732n
(11) Shimizu,M.;Mochida,K.;Asai,Y.;Yamatani,A.;Kaki,R.; Hiyama,T.;Nagai,N.;Yamagishi H.;Furutani,H.J.Mater. Chem.2012,22,4337.doi:10.1039/c2jm15173a
(12)Yamaguchi,S.;Xu,C.;Tamao,K.J.Am.Chem.Soc.2003,125, 13662.doi:10.1021/ja038487+
(13)Xu,C.;Yamada,H.;Wakamiya,A.;Yamaguchi,S.;Tamao,K. Macromolecules 2004,37,8978.doi:10.1021/ma0486546
(14)Mouri,K.;Wakamiya,A.;Yamada,H.;Kajiwara,T.; Yamaguchi,S.Org.Lett.2007,9,93.doi:10.1021/ol062615s
(15)Xu,C.;Wakamiya,A.;Yamaguchi,S.J.Am.Chem.Soc.2005, 127,1638.doi:10.1021/ja042964m
(16) Kaszynski,P.;Dougherty,D.A.J.Org.Chem.1993,58,5209. doi:10.1021/jo00071a034
(17) Patil,S.A.;Scherf,U.;Kadashchuk,A.Adv.Funct.Mater.2003, 13,609.doi:10.1002/(ISSN)1616-3028
(18)Wakim,S.;Bouchard,J.;Blouin,N.;Michaud,A.;Leclerc,M. Org.Lett.2004,6,3413.doi:10.1021/ol048543r
(19)Kawaguchi,K.;Nakano,K.;Nozaki,K.J.Org.Chem.2007,72, 5119.doi:10.1021/jo070427p
(20) Su,H.C.;Fadhel,O.;Yang,C.J.;Cho,T.Y.;Fave,C.;Hissler, M.;Wu,C.C.;Reau,R.J.Am.Chem.Soc.2006,128,983.doi: 10.1021/ja0567182
(21)Baumgartner,T.;Neumann,T.;Wirges,B.Angew.Chem.Int. Edit.2004,43,6197.doi:10.1002/(ISSN)1521-3773
(22)Baumgartner,T.;Bergmans,W.;KmrpMti,T.;Neumann,T.; Nieger,M.;NyulMszi,L.Chem.Eur.J.2005,11,4687.doi: 10.1002/(ISSN)1521-3765
(23) Dienes,Y.;Durben,S.;KmrpMti,T.;Neumann,T.;Englert,U.; NyulMszi,L.;Baumgartner,T.Chem.Eur.J.2007,13,7487. doi:10.1002/(ISSN)1521-3765
(24) Fukazawa,A.;Hara,M.;Okamoto,T.;Son,E.-C.;Xu,C.; Tamao,K.;Yamaguchi,S.Org.Lett.2008,10,913.doi: 10.1021/ol7030608
(25) Han,L.Z.;Wang,Z.;Hua,Y.J.;Ren,A.M.;Liu,Y.L.;Liu,P. J.Acta Chim.Sin.2012,70,579.[韩立志,王 卓,华英杰,任爱民,刘艳玲,刘朋军.化学学报,2012,70,579.]doi: 10.6023/A1106061
(26) Chen,L.T.;Yan,Y.;Zhang,C.;Ma,C.A.Acta Chim.Sin.2010, 68,2167.[陈丽涛,严 妍,张 诚,马淳安.化学学报, 2010,68,2167.]
(27) Sun,H.T.;Tian,X.H.;Yuan,Y.Z.;Sun,J.Y.;Sun,Z.R.;Zhuo, X.L.Acta Phys.-Chim.Sin.2011,27,1847.[孙海涛,田晓慧,元以中,孙金煜,孙真荣,卓小玲.物理化学学报,2011,27, 1847.]doi:10.3866/PKU.WHXB20110840
(28) Liu,X.J.;Lin,T.;Gao,S.W.;Ma,R.;Zhang,J.Y.;Cai,X.C.; Yang,L.;Teng,F.Acta Phys.-Chim.Sin.2012,28,1337. [刘小君,林 涛,高少伟,马 睿,张晋悦,蔡新晨,杨 磊,滕 枫.物理化学学报,2012,28,1337.]doi:10.3866/PKU. WHXB201204092
(29)Yue,Y.;Xu,H.X.;Hao,Y.Y.;Xie,X.D.;Qu,L.T.;Wang,H.; Xu,B.S.Acta Phys.-Chim.Sin.2012,28,1593.[岳 岩,许慧侠,郝玉英,解晓东,屈丽桃,王 华,许并社.物理化学学报,2012,28,1593.]doi:10.3866/PKU.WHXB201204181
(30)Wang,F.J.;Zhou,D.H.;Zuo,S.Y.;Cao,J.F.;Peng,X.J.Acta Phys.-Chim.Sin.2012,28,1645.[王凤娇,周丹红,左士颖,曹建芳,彭孝军.物理化学学报,2012,28,1645.]doi:10.3866/ PKU.WHXB201205083
(31) Zhao,P.;Xu,L.C.;Ma,L.Acta Phys.-Chim.Sin.2011,27, 2541.[赵 平,绪连彩,马 丽.物理化学学报,2011,27, 2541.]doi:10.3866/PKU.WHXB20111021
(32) Keruckas,J.;Lygaitis,R.;Simokaitiene,J.;Grazulevicius,J.V.; Jankauskas,V.;Sini,G.J.Mater.Chem.2012,22,3015.doi: 10.1039/c2jm14387a
(33) Shi,L.L.;Hong,B.;Guan,W.;Wu,Z.J.;Su,Z.M.J.Phys. Chem.A 2010,114,6559.doi:10.1021/jp1010617
(34) Liu,T.;Xia,B.H.;Zheng,Q.C.;Zhou,X.;Pan,Q.J.;Zhang, H.X.J.Comput.Chem.2010,31,628.
(35)Yin,J.;Chen,R.F.;Zhang,S.L.;Ling,Q.D.;Huang,W. J.Phys.Chem.A 2010,114,3655.
(36) Liu,S.J.;Sun,S.;Wang,C.M.;Zhao,Q.;Sun,H.B.;Li,F.Y.; Fan,Q.L.;Huang,W.ChemPhysChem 2011,12,313.doi: 10.1002/cphc.v12.2
(37) Marcus,R.A.J.Chem.Phys.1956,24,966.doi:10.1063/ 1.1742723
(38)Kan,Y.H.;Wu,K.;Zhu,Y.L.;Hou,L.M.;Su,Z.M.Acta Phys.-Chim.Sin.2010,26,1423.[阚玉和,吴 凯,朱玉兰,侯丽梅,苏忠民.物理化学学报,2010,26,1423.]doi:10.3866/ PKU.WHXB20100528
(39)Sun,G.Y.;Li,H.B.;Geng,Y.;Duan,Y.A.;Tang,X.D.;Su,Z. M.;Zhou,Z.Y.Chem.J.Chin.Univ.2012,33,794.[孙光延,李海斌,耿 允,段雨爱,汤肖丹,苏忠民,周子彦.高等学校化学学报,2012,33,794.]
(40) Hutchison,G.R.;Ratner,M.A.;Marks,T.J.J.Am.Chem.Soc. 2005,127,2339.doi:10.1021/ja0461421
(41) Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian 03, Revision E.01;Gaussian Inc.:Wallingford,CT,2004.
(42) Lee,C.;Yang,W.;Parr,R.G.Phys.Rev.B 1988,37,785.doi: 10.1103/PhysRevB.37.785
June 25,2012;Revised:August 27,2012;Published on Web:August 27,2012.
Optoelectronic Properties for Main Group Element-Bridged Ladder Compounds
LIU Shu-Juan†MA Ting-Chun†XU Wen-Juan LIU Xiang-Mei ZHAO Qiang*HUANG Yan-Qin HUANG Wei
(Key Laboratory for Organic Electronics&Information Displays,Institute of Advanced Materials, Nanjing University of Posts&Telecommunications,Nanjing 210046,P.R.China)
Ladder-type π-conjugated molecules with fully ring-fused structures have fascinating optoelectronic properties because the flattened π-conjugated framework can eliminate conformational disorder and effectively enhance π-conjugation.Their optoelectronic properties can be modified by incorporating main group elements into the ladder skeleton.Heteroatom-bridges not only stiffen the skeleton but also contribute to the electronic structure through orbital interaction between the main group elements and the π-conjugated skeleton.Herein,the structural,electronic,and optical properties of bisand tetrakis-bridged(C,Si or P-bridged)stilbene derivatives were investigated by density functional theory (DFT)and time-dependent DFT(TDDFT)to provide theoretical understanding and predictions for these compounds.The electronic structures of these π-conjugated skeletons could be tuned by the incorporated elements.Compared with bis-bridged analogs,tetrakis-bridged derivatives exhibited substantial red shifts in the absorption and shorter radiative lifetimes because of extended π-conjugation.In addition,the energy barrier for the injection and transport rates of the holes and electrons was evaluated using ionization potentials,electronic affinities,and reorganization energies(λ).Compared to bis-bridged analogs, tetrakis-bridged derivatives exhibit higher accepting abilities for both holes and electrons.
Density functional theory; Electronic property; Ladder-type molecule; Main group element; Photophysical property
10.3866/PKU.WHXB201208272
O641
∗Corresponding author.Email:iamqzhao@njupt.edu.cn;Tel/Fax:+86-25-85866396.
†These authors contributed equally to this work.
The project was supported by the National Key Basic Research Program of China(973)(2009CB930601,2012CB933301),National
Natural Science Foundation of China(21174064,21171098),the Ministry of Education of China(IRT1148),Key Projects in Jiangsu Province for International Cooperation,China(BZ2010043),Nanjing University of Posts and Telecommunications,China(NY210029),and Priority
Academic Program Development of Jiangsu Higher Education Institutions,China.
国家重点基础研究发展计划(2009CB930601,2012CB933301),国家自然科学基金(21174064,21171098),教育部创新团队(IRT1148),江苏省国际合作计划-重点国别和地区研发合作项目(BZ2010043),南京邮电大学基金(NY210029)和江苏高校优势学科建设工程资助项目资助
