云杉属树种天然群体DNA标记的国外研究进展
2012-11-28罗建勋辜云杰
罗建勋,董 昕,辜云杰
(1.四川省林业科学研究院,四川成都 610081;2.四川农业大学林学院,四川雅安 625014)
云杉属(Picea Dietr.)植物历史悠久,全世界约有40种[1]。据古气候学和古环境学研究证实,云杉属植物早在白垩纪就在地球上有分布,后经地质变迁,形成了现今的分布格局,即主要分布在北纬50°~60°的寒温带或冻原地带,少数分布在温带和亚热带的湿冷亚高山带上。云杉属植物因其树干通直圆满,材质好,出材率高,而且适生性强,现已成为西欧、北欧、波罗的海沿岸国家、俄罗斯和加拿大的重要造林树种与工业用材林树种。同时,云杉属树种高度异交,杂合度较高,其天然群体具有丰富的遗传多样性,所以是研究遗传变异的好材料,通过发掘变异和利用遗传变异,达到林木遗传改良和种质资源保存的目的。迄今为止,对云杉属遗传变异的研究先后经历了形态学标记、细胞学标记、同工酶标记和DNA标记4个阶段,目前以DNA分子水平的研究为热点。DNA标记技术起源于20世纪70年代的西欧,而后迅速发展,被各地广泛应用,尤其是21世纪头10a,该项技术更加推动了国外云杉属植物遗传多样性研究在分子水平上的发展,并取得了卓有成效的成果。然而,我国云杉资源丰富,但DNA分子标记方面的研究起步较晚,有待进一步发展。就论文发表情况而言,仅在1990年~2000年间,曾见罗建勋[2]、张含国[3]、王芋华[4]等少数几个人对国外云杉遗传多样性进行过详细论述。综上,有必要对2000年~2010年国外云杉属植物天然群体的DNA标记进行综述。
1 DNA标记在云杉天然群体遗传研究中的应用
DNA分子标记技术已经在云杉属树种的遗传多样性分析、遗传图谱构建、种的鉴定与亲缘关系分析和基因表达分析等方面得到了广泛的应用,尤其是在遗传多样性与遗传分化分子水平的研究必不可少,已经从核DNA研究扩展到覆盖线粒体DNA(mtDNA)和叶绿体 DNA(cpDNA)的研究。例如Maghuly等[5]开发了挪威云杉(Picea abies)群体的线粒体遗传标记。众所周知,种群是物种进化的基本单位,群体在自然界中有其特定的分布格局式样,所以研究遗传多样性不仅要分析遗传变异的高低,还要了解遗传变异在群体内和群体间的分布式样以及在时间上的变化,即遗传结构。然而,云杉属树种的基因组较大,2C=29 ×109bp[6],且 70%是非编码区,基因的表达尚不清楚,对基因组的量化描述也存在局限性,因此,利用DNA标记对云杉属的变异进行定量分析,可以追寻其进化规律的踪迹,从而能更好地利用现存云杉遗传材料。根据各种标记的开发基础,主要将其分为4类。
1.1 基于DNA-DNA杂交的分子标记与云杉天然群体的遗传研究
1.1.1 RFLP标记在云杉天然群体遗传研究中的应用
RFLP(restriction fragment length polymorphism,限制性片段长度多态性)是第一个被应用于遗传研究的 DNA分子标记,1974年由 Grozdicker创立。RFLP是一种共显性遗传标记,首先利用限制性内切酶将基因组DNA消化成不同分子量的同源片段,而后经电泳分离再转移到滤膜上,然后进行southern杂交,最后通过放射性自显影或酶学检测技术检测限制性酶切片段多态性。Dering和Lewandowski[7]为了确定冰河后期挪威云杉从避难所迁出形成的接合区域,利用PCR-RFLP标记分析了58个挪威云杉群体1 353个个体的线粒体基因nad1 b/c多态性,检测到波兰境内的挪威云杉有两种DNA变异型,即“南部单体型”和“北部单体型”。研究发现这些单体型的地理分布与特有的挪威云杉分布范围和避难所起源相符。“北方型”主要分布在挪威云杉分布区的北部,“南方型”主要分布在挪威云杉分布区的南部。两种单体型同时出现在无云杉区域(根据花粉数据推测,在挪威云杉天然分布区内以中欧为界分成南北两个区域,在中欧的断裂处形成南部与北部的重叠分布区,也叫无云杉区),无云杉区域内,以标记为“南方单体型”的群体出现的频率较高。结合化石和花粉数据认为,波兰中部平原的挪威云杉群体分别从阿尔巴千山和俄罗斯迁移至此,而后交汇形成。起源于阿尔巴千山的云杉在重叠区域分布居多,可能是因为该起源比俄罗斯起源迁移得更早。
1.1.2 VNTR标记在云杉天然群体遗传研究中的应用
VNTR(variable number of tandem repeats,数目可变串联重复序列)也叫小卫星标记,是共显性遗传标记,由几个到几十个核苷酸序列重复构成,拷贝数为几十到几百不等。VNTR的原理与AFLP的基本相同,但要求VNTR的酶切位点在重复序列之外,且内切酶在基因组的其他部位有较多酶切位点,同时还需满足杂交DNA探针序列与微卫星或小卫星序列相同。尽管小卫星标记的多态信息量较高,但其在基因组中分布不均匀,因此,不如其他标记应用范围广,在云杉群体多样性研究中也仅有少量的报道。Bastien等[8]以自然分布区内的14个挪威云杉群体92个个体为材料,检测其mtDNA多态性,共获得11条长度为131-447bp的多态性序列,这些序列由3个串联基序以不同拷贝数重复形成。串联重复数的差异反映出群体间基因多态性很高,群体间分化很大,遗传分化系数Gst=0.749。
1.2 基于PCR技术的分子标记与云杉天然群体的遗传研究
1.2.1 RAPD标记在云杉天然群体遗传研究中的应用
RAPD(randomly amplified polymorphic DNA,随机扩增多态性DNA)是由Williams于1990年发明的一种检测核苷酸序列多态性的方法。RAPD通常用1个8 bp~10 bp随机引物非定点地扩增基因组DNA,然后利用PCR技术从扩增的DNA片段上分析多态性,即反映出不同遗传材料的多态性。因这种标记技术要求低,对于生物学资料未知的基因组就可检测到大量多样性信息,加之成本低,所以几乎用于云杉属的各种遗传分析。Collignon等[9]在挪威云杉的整个自然分布区内对其进行RAPD和数量性状的地理变异分析。据此,挪威云杉被划分成北欧和中欧两个地理上的类群,每个类群内的数量性状和DNA之间存在着明显的解偶联。然而,将分子方面和数量性状方面提供的信息结合起来能够为地理变异模式提供新的深入了解:发现在Baltico-Nordic区域是以纬度梯度为主导的地理变异模式,这与通过花粉数据推断的云杉以东-西方向迁移为主导的地理变异模式明显相悖;然而,在中欧地区,云杉以经度梯度为主导的迁移与花粉报告的结果一致。该研究对于通过数量性状和DNA得到的一致和矛盾的结果,以挪威云杉发生历史性事件的形式对其进行了讨论。Jeandroz等[10]以法国境内8个挪威云杉群体(6个天然群体,1个种子园群体,1个栽植群体)为材料应用RAPD和mtDNA标记对挪威云杉,进行了群体间的变异研究。研究通过RAPD表型分析确定了法国境内欧洲云杉遗传资源的3个群体,它们分别是孚日山脉(天然源群体和栽植群体)群体、北阿尔卑斯山群体或侏罗纪山脉群体以及南阿尔卑斯山群体。境内群体的基因多样性值差异不大。线粒体标记的研究结果显示,在DNA MH44位点处存在一个重要变异,6个孚日山脉的天然参试群体表现出阿尔卑斯山脉群体特有的特征。孚日山脉的栽植群体表现出两个额外的mtDNA类型,这支持了19世纪后期法国境内的欧洲云杉是从欧洲东部迁移而来的假说。
1.2.2 SCAR标记在云杉天然群体遗传研究中的应用
SCAR(sequence-characterized amplified region,序列特征化扩增区域)是将所找到的RAPD标记进行克隆并分析,根据RAPD两侧序列设计特异引物,再对基因组DNA进行PCR扩增而产生的分子标记。SCAR常以扩增产物的有无反映被检DNA的差异性,此时为显性标记;有时也表现为扩增片段长度的多态性,此时为共显性标记。Scotti等[11]在研究挪威云杉上一次冰期后再侵入阿尔卑斯山的迁移路线时,曾对沿阿尔卑斯山脉的7个群体和一个亚平宁山脉群体的7个SCAR位点进行特征分析,发现该区域内挪威云杉的遗传变异分布不均匀,群体间遗传分化程度较高,遗传分化系数Fst=0.118。根据分子方差分析和距离分离模型检验结果,在亚平宁山脉存在冰期种源区的假说不成立,认为亚平宁山脉群体在地理上处于冰期后再次侵入群体分布区的边缘,是由迪纳拉山脉群体从东北向西南迁移至此形成的,而邻海的阿尔卑斯山脉则可能有残余群体,其挪威云杉群体被南北方向的分界线划分成两部分。尽管SCAR标记在云杉群体遗传多样性研究中的应用不如RAPD标记广泛,但其能够弥补RAPD产生假带、不稳定的不足,在云杉属树种的亲缘关系分析中发挥了一定作用。
1.2.3 AFLP标记在云杉天然群体遗传研究中的应用
AFLP(Amplified Fragment Length Polymorphism,扩增片段长度多态性)是RAPD标记和RFLP标记的组合,根据限制性内切酶酶切片段的不同长度检测基因多态性,同时兼有RAPD标记的简便性和RFLP标记的可靠性,被认为是一种先进的DNA分子标记,即第二代分子标记。AFLP标记被广泛用于云杉属树种的遗传多样性分析、遗传图谱构建和基因定位等。XR Wang等[12]以瑞典北部7个岛(有的岛发生过林火,且岛的面积不同)上的挪威云杉群体共117个个体进行群体结构的研究,利用105个AFLP多态性标记对材料分析,获得96个特异的基因型,平均基因多样性P为0.37,群体间遗传变异高,群体间遗传分化系数Fst=0.19。研究认为遗传漂变、奠基者效应、群体小、低频率的有性生殖以及产种量少是导致群体间遗传变异高的原因。
1.2.4 SSR标记在云杉天然群体遗传研究中的应用
SSR(Simple Sequence Repeat,简单序列重复)标记又叫微卫星DNA,通常由1~6个核苷酸组成的核心序列串联重复而成,长度通常不超过200bp,因其两侧序列为相对保守的单拷贝序列,可根据两侧序列设计特异引物,经PCR扩增获得SSR的核心序列,由于核心序列的串联重复数不同,所以PCR产物长度不同,进而揭示SSR的多态性。SSR因具有共显性、多态性丰富等优点,所以是群体遗传结构和多样性研究、遗传图谱构建等的理想工具。Hodgetts等[13]开发了白云杉(Picea glauca)的 SSR标记,利用13对引物扩增加拿大西部3个地区的白云杉材料,获得14个多态性位点,每位点等位基因数为3~32不等,观测杂合度Ho为0.33~0.94不等,说明白云杉SSR标记具有很高的遗传多态性。用相同的引物去扩增黑云杉(P.mariana)、红云杉(P.rubens)、挪威云杉(P.abies)、科罗拉多云杉(P.pungens)、北美云杉(P.sitchensis)和恩氏云杉(P.engelmannii),大多数都能扩增出多态性位点,且多态性较高;用这些引物去扩增非云杉属的针叶树种,发现并不是所有的都能得到多态性产物,说明这些标记可能仅限于云杉属树种拥有。A’Hara和Cottrell[14]以一些无亲缘关系的北美云杉(Picea sitchensis)和其全同胞家系子代为材料,进行基因组微卫星分析得到观测杂合度Ho为0.38~0.91,每位点等位基因数A为6~21,平均每位点等位基因数为12.2,并得出其全同胞家系子代多态性位点分离比遵守孟德尔遗传规律的结论。Scotti等[15]为了粗略获得挪威云杉染色体组结构图谱,以瑞典4个不同区域25年生的85个杂交子代个体为材料,利用6种(AFLP,SSR,S-SAPs,IRAP,ESTP 和 inter-LTR)不同的分子标记建立了模拟测交遗传连锁图谱。该研究利用3种以上的标记分别获得了27个母本图谱的连锁群和23个父本图谱的连锁群,根据不同标记在两个图谱上的分布特点将二者整合,得到具有13个连锁群的双亲图谱,再以此图谱检测每种标记在染色体中出现的频率,并利用自相关分析找到彼此关联的标记,确定他们在染色体上的分布情况。研究发现不同序列单元在基因组中分布不均匀,有些标记聚集分布,而有些有关联的序列却随机分布,研究还发现并不是所有的标记都在染色体上具有等量的连锁群,说明挪威云杉染色体空间结构不是均质的。Bastien[8]应用微卫星标记对长为11bp的变异序列进行了系统发生分析,建立的系统发育树很好地展示了群体间亲缘演化关系。Nasri等[16]利用父系遗传的叶绿体微卫星研究地理上严格受限的塞尔维亚云杉地方树种P.omorika的群体遗传结构。利用7个天然群体,每群体14个个体扩增出9个cpSSR片段,其中包括5个多态性区域,获得4个不同的单体型。平均总单体型多样性HT=0.395,平均群体内多样性HS=0.279,其单体型变异是目前描述过的大多数针叶树中较低的,并依据单体型变异将其划分为南部和北部2个地理群体。北部群体仅有一种单体型,而南部群体表现为2种~3种单体型。研究认为当前形成的P.omorika群体遗传结构是由第四纪冰期的瓶颈作用和遗传漂变刻画的。因此,认为P.omorika是遗传衰退树种中的一个小群体。Ballian等[17]以巴尔干半岛的13个群体(其中2个群体与Nasri选用的一样)为材料进行研究,结果表明:尽管群体间遗传分化明显,遗传分化系数Fst=0.261,但未检测到遗传变异的地理分化。群体结构的贝叶斯分析结果显示,当前的塞尔维亚云杉是其祖先P.omoricoides遗留下的一个分支。Anderson等[18]对横贯北美东北部的24个白云杉群体的cpDNA进行测序,发现大多数单体型是特异的,在阿拉斯加区域的单体型多样性相当高,显然白云杉很久以前就在具有极端气候冰河期的异质生境中存在,这一结果反驳了白云杉在冰河后期由南部的劳伦太德冰盖向北部迁移的结论。这些结果都表明,从化石记录中推测的迁移速率太高,白云杉适应气候变化的能力不如之前想象的那样强。Aizawa等[19]以鱼鳞云杉(Picea jezoensis)mtDNA 和 cpDNA为材料,对日、俄、中、韩共33个天然群体的遗传结构进行分析,描绘了鱼鳞云杉在日本周围海峡交汇处的线粒体单体型地理分布模式(群体间遗传分化系数GST=0.901;基因流NST=0.934)。然而,以前的鱼鳞云杉群体叶绿体单体型分布模式资料显示日本周边海峡处花粉流数据与此差异很大(GST=0.233;NST=0.333)。物种的地理隔离成为种子和花粉迁移的障碍,隔离的群体分别独立进化,在群体交汇处则表现出明显的遗传分化。因为勘察加半岛(位于苏联北部)群体不相连接,所以推测勘察加半岛是鱼鳞云杉当前分布的北缘。研究发现,在日本境内的北海道岛和本州岛之间存在鱼鳞云杉的地理变异型。就此讨论了东北亚云杉的演化历史,认为本州岛的群体可能是从亚洲大陆经朝鲜半岛迁移过去,北海道群体可能从亚洲大陆经库页岛迁移过去。日本境内两岛上的群体具有特有线粒体单体型,可能是因第四纪冰河后期两岛与亚洲大陆在地理上的隔离所致。
1.2.5 ISSR标记在云杉天然群体遗传研究中的应用
ISSR(Inter-simple Sequence Repeat,简单序列重复区间)标记由Zietkiewicz等[20]于1994年开发,是一种用于检测SSR区间序列差异的分子标记技术。与SSR标记相比,ISSR标记不需要已知微卫星两侧序列去设计引物,就可扩增出目的片段,稳定性好,且能比RFLP、RAPD和SSR标记反映出更多的多态性信息,在云杉属群体遗传多样性分析中应用得较好。Sylwia Dobrzeniecka等[21]利用 ISSR 标记对不同重金属污染程度的高地和洼地上黑云杉群体进行遗传分析。多态位点百分率P为65%~90%不等,平均多态位点百分率为75%;平均每位点等位基因观测数Na为1.650~1.900,平均有效等位基因数Ne为1.168~1.632;群体内遗传多样性h为0.264~0.359不等,平均群体内遗传多样性为0.310;香浓多样性指数I为0.381~0.524,平均值为0.449;群体遗传距离为0.171~0.351。研究表明,黑云杉群体具有较高的总遗传变异,但群体间的遗传变异很低,该研究认为群体内的遗传变异很高。该结论与 Perry和 Bousquet[22]的研究相符。而 Rajora和Pluhar[23]在马尼托巴省的的东部和北部区域同样对黑云杉天然群体的多样性进行分析,并没有发现群体间存在显著变异,就此认为黑云杉群体间的遗传多样性不受地理因素的影响。ISSR可与其他标记联合用于云杉属树种的鉴定。K.K.Nkongolo等[9,10]应用 RAPD、ISSR 分子标记鉴定了红云杉、白云杉、黑云杉和恩氏云杉。该研究用ISSR的827号引物分别扩增出450 bp的恩氏云杉特异性片段和770 bp的白云杉特异性片段;用ISSR的841号引物分别扩增出230 bp的白云杉特异性片段和470 bp的黑云杉特异性片段;同样根据RAPD标记获得的红云杉和黑云杉特异性片段将4种云杉鉴别出来。幼龄时期的黑云杉在形态上与白云杉和红云杉相似,利用分子标记分析可将黑云杉从红云杉和白云杉中鉴别出来。研究采用ISSR标记和SCAR标记进行分析,认为ISSR分析中270 bp片段是白云杉的特征性标记,470bp片段为黑云杉的特征性标记,SCAR分析中792 bp片段为红云杉的特征性标记,从而,根据 ISSR和 SCAR分析将黑云杉筛选出来[21]。
1.3 基于mRNA的分子标记与云杉天然群体的遗传研究
1.3.1 EST标记在云杉天然群体遗传研究中的应用
EST(Expressed sequence tags,表达序列标签)是长为150 bp~500 bp的基因表达序列片段。EST标记是将mRNA反转录成cDNA并克隆到载体构建成cDNA文库,再大规模随机挑选cDNA克隆,对其5'端或3'端进行一步法测序,将所获序列与基因数据库已知序列比较,从而获知生物体生长发育、繁殖分化、遗传变异、衰老死亡等一系列生命过程。EST被看作是获得重要基因序列的快捷途径,近年来,开发出一系列适用于植物研究的EST标记,如ESTSSR、EST-RFLP、EST-AFLP和 EST-SNP等。这些标记在云杉属植物的遗传研究中得到了高效的应用。EST-SSR分子标记是基于表达序列标签开发微卫星的一种新型分子标记。由于云杉的基因组比较大,重复的DNA序列较多,经常产生复杂的多位点扩增产物,使 SSR标记在云杉的应用中受到限制,而EST-SSRs标记在这一点比SSR标记具有优越性。EST-SSR与基因组SSR相比,具有可在植物物种之间转移的优点。目前,EST-SSR被广泛应用于植物基因组学方面的研究,如遗传图谱构建、比较作图、遗传多样性评价、种质鉴定、系统发育与进化研究等[24-29]。Silvia Fluch 等[30]通过14 022个挪威云杉EST序列建立SSR标记并对其进行特征分析,发现三核苷酸重复基序在数据集中出现最多,五核苷酸和六核苷酸重复基序次之;用特异性引物对扩增60个位点,检测到测试群体(16个个体)和筛选群体(96个个体)中有27个位点是多态的。每位点等位基因数A为2~17个,观测杂合度Ho为0.075~0.99。该研究检测到筛选群体中具有很高的遗传变异,因此认为EST-SSR标记适用于挪威云杉群体功能基因的变异分析。Yanik Bérubé等[31]曾对云杉的EST数据库进行了EST-SSRs特征描述,根据白云杉,恩氏云杉及北美云杉的数据分析获得629个云杉的特异标记。A'Hara和 Cottrell[14]将利用北美云杉gSSR检测到的遗传参数与在云杉EST文库中利用微卫星标记获得的遗传参数进行比较,结果表明,gSSR检测到的每位点等位基因数高于他们之前通过 EST-SSR 检测[32]到的结果。Rungis等[33]的研究也得出了相同的结论。
1.3.2 STS标记在云杉天然群体遗传研究中的应用
STS(Sequence-tagged Site,序列标记位点)是在基因组中染色体特定位置上唯一出现的一段已知序列,可以作为界定基因组中染色体片段位置的位标[34]。STS标记兼具RAPD标记的简便性和 SSR标记的特异性,在云杉群体遗传分析中得到了应用。Bennuah等[35]对英国西北部哥伦比亚北美云杉和白云杉基因渐渗区的16个区域57个家系种子和幼苗的684个个体进行遗传分析,检测其STS等位基因频率,平均STS杂合指数为0.46~0.95。研究表明群体间遗传分化显著,且群体杂合指数从沿海向陆地逐渐降低,表现出地理上的变异。杂合指数的多重回归分析显示地理间距主导群体间遗传分化。Perry和 Bousquet[22]应用12个STS标记比较黑云杉群体遗传多样性和交配系统在火烧前、后的差异,结果表明3个原迹地群体和4个火烧迹地群体的遗传杂合度、等位基因数量、固定指数都分别相近,群体间遗传分化不显著。交配系统指数在火烧前后也无显著变化,没有发现远缘杂交种。因此认为迹地上黑云杉并没有因林火的影响降低遗传多样性,交配系统也比较稳定。Gapare等[36]对北美云杉8个群体的遗传多样性进行研究。根据其地理分布特点分为核心群体和边缘群体,对北美云杉群体进行STS分析,结果表明,核心群体的平均期望杂合度与边缘群体的极为相似,分别为0.58和0.56;但二者近交系数相差很多,分别为0.03和0.17;核心群体和边缘群体的基因流分别为9.0和3.5。8个群体中,75%以上的等位基因为常见的广域基因,只有一个为稀有的局域基因,仅分布于一个不与其他群体相连的核心群体和两个不相连的边缘群体。研究证明,在不相连的外围群体内比相连的核心群体内发生过的瓶颈作用强烈。因此,建议优先对不相连的边缘群体进行原地保存或扩大样本进行异地保存,可将稀有基因保存下来。Gapare[37]在之前的北美云杉群体空间遗传结构研究中,曾对8个核心群体和边缘群体每群体200个个体进行空间作图和基于cDNA的STS分子标记分析。对一定间距内个体间的平均共祖系数检测结果显示:在核心群体中,距离50 m的个体间等位基因和基因型的分布是随机的,共祖值不显著,而边缘群体中,相距小于50 m的个体间有相似的多态性位点基因型,共祖系数比核心群体的高0.20;当间隔达到500 m时,共组系数还为正值。研究认为,核心群体繁殖个体的密度相对大,可能导致种子互相覆盖,制约了空间遗传结构的发展,而边缘群体中繁殖个体密度相对较小,由于子代在母树很近的地方繁殖,后来又发生近亲繁殖,很多群体在更新世后期的树种扩张区域再繁殖,所以等位基因和基因型的分布更具结构性。在制定基因保存策略时,可能需要对更多的边缘群体种质进行原地保存。研究中有关空间遗传结构的数据可为育种策略的制定和种子资源收集调查提供依据。
1.3.3 RT-PCR标记在云杉天然群体遗传研究中的应用
RT-PCR(reverse transcription-PCR,逆转录PCR)是将一条RNA链逆转录成互补的DNA,再以互补的DNA为模板通过PCR进行扩增。该标记的实用性在于可对基因表达进行实时监测。Holliday等[38]对具有21 840个特异元件的3个北美云杉群体cDNA微阵列进行群体内和群体间秋季基因表达监控。利用该标记可以检测云杉微阵列芯片数据,对其基因表达进行监测,然后通过RT-PCR验证候选基因的微阵列芯片数据。研究表明,脱水蛋白、与发病机制相关或防冻相关的基因,碳水化合物和脂代谢基因以及信号转到和转录调控相关的基因数量在秋季有所增加。群体间微阵列芯片的早秋和晚秋杂交结果揭示了秋季转录体的基本变异,认为一些时间点上的杂交还能反映出对环境的局部适应,为研究北美云杉耐冷机制提供备了资料。
1.4 基于单核苷酸多态性的分子标记与云杉天然群体的遗传研究
SNP(Single Nucleotide Polymorphism,单核苷酸多态性)标记是个体间发生的最普遍的遗传变异形式,在基因组中分布广泛,比SSR标记密度还要大,平均每1 000个碱基对中就会有一个碱基发生变异[39]。加之其遗传稳定性很高,具有易实现分析和自动化等优点,已成为继RFLP和SSR标记之后的第三代DNA分子标记,渗透到了云杉属的遗传多样性分析和联合作图等方面的研究。Namroud等[40]对6个白云杉天然群体进行SNP分析,从345个表达基因中检测到534个SNPs,平均期望杂合度He为0.270,遗传分化系数Gst为0.006。研究认为是自然选择导致白云杉发生了基因分化。Holliday等[41]以14个北美云杉群体410个个体为材料,检测与发芽调控和秋季耐寒有关的基因多态性并进行关联作图,从200多个表达基因中检测到768个SNPs。Q聚类和R聚类后,分析28个候选基因的关联性,发现这些候选基因与耐寒表型变异和发芽调控表型变异相关程度分别为28%和34%;推测存在5个与光信号转导关联的重要基因。研究还发现,很多与表型关联的SNP至少与一种气候变化相关。
2 不同DNA标记检测云杉属树种所获遗传参数的比较与分析
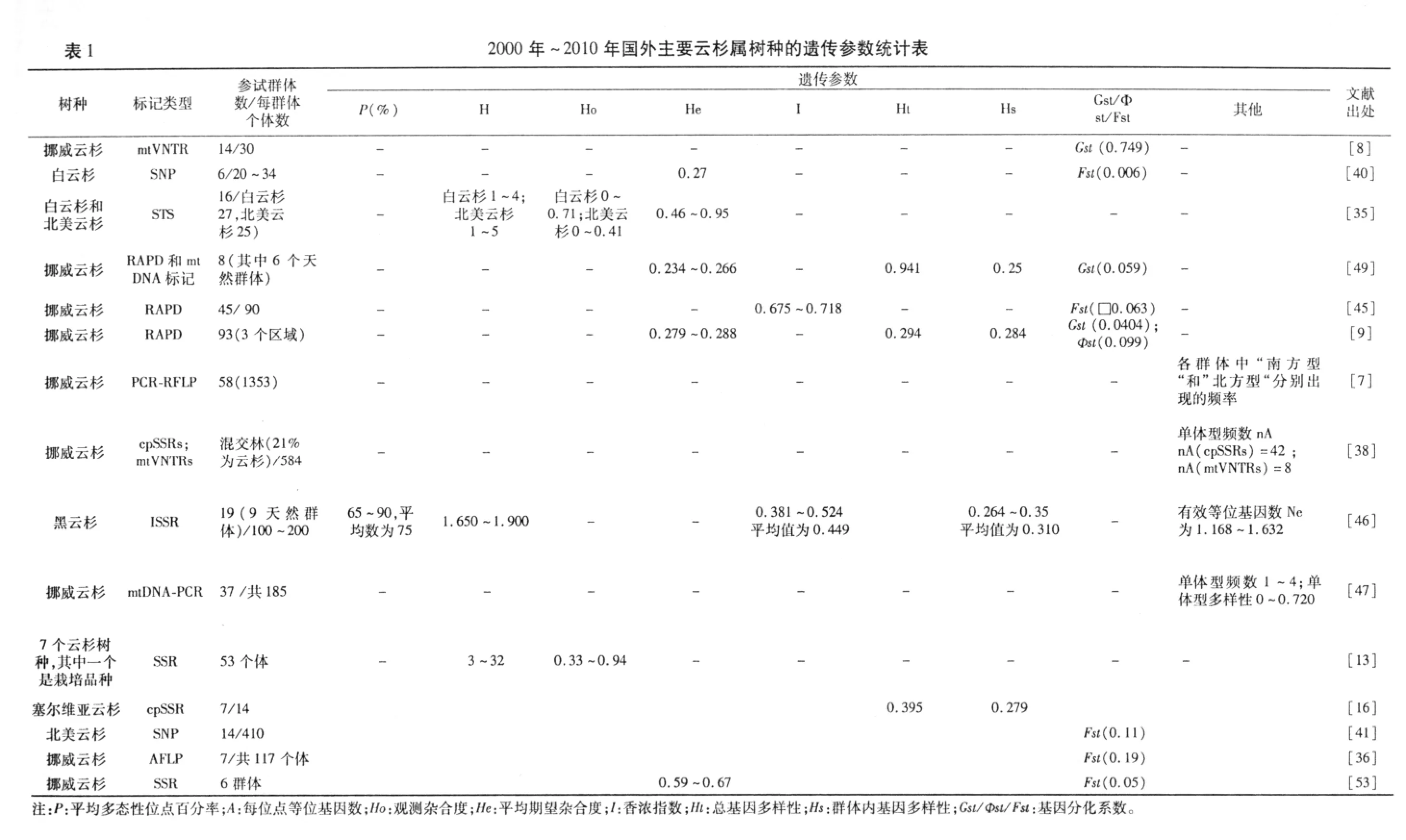
?
在评价群体遗传多样性水平时,通常涉及到5个遗传参数:平均多态位点百分率(P)、平均期望杂合度(He)、平均Shannon指数(I)、Nei's遗传分化值(Gst)和基于AMOVA分析的遗传分化值(Фst)。其中,Gst和Фst为分化指数Fst的近似值。20世纪80年代,Ayala[42]研究组对主要动植物的遗传多样性研究进行了总结,认为P和He是衡量遗传多样性水平的重要参数。随后,Hamrick[43]研究组的统计结果支持了Ayala等人的报道。而Qian等[44]认为参数He和I在遗传多样性的评估上比P更加有效。近年来,较多遗传多样性研究都借助于分子标记的手段,多样性的分析和评估主要参照Hamrick研究组的统计结果。通过对利用不同标记所获云杉的遗传参数比较分析,具体见表1,认为云杉天然群体遗传变异丰富,对环境变化适应能力强,几乎所有研究表明,群体间存在遗传分化,但遗传分化系数并不高。对相同云杉属树种的遗传多样性及遗传分化分析时,由于参试群体数和个体数不同,加之采用的DNA标记类型不同,得出的结论不尽相同。
因为不同类型的分子标记探测范围不同,检测多态性的程度不同,所以,分析相同材料所获遗传参数值表现出差异,甚至得出相反的结论。Namroud等[48]在扫描白云杉单核苷酸多态性基因组的研究中对几种不同的分子标记进行了比较,该研究通过SNP扫描得到的期望杂合度均低于通过等位酶、RAPD、AFLP、SSR和 ESTP得到的杂合度,原因是SNP检测双等位基因,而且等位基因突变率很低,加上植物编码区域本来就比非编码区域的核苷酸多态性低,所以得到的多态位点百分率低,而期望杂合度是通过多态位点百分率计算而得,所以采用SNP获得的期望杂合度比采用其他几种检测手段检测到的遗传多样性参数值低。Jeandroz等[49]对6个挪威云杉天然群体的同一套材料,分别应用RAPD和mtDNA标记进行检测,RAPD的分子方差分析结果显示6个群体差异显著,而用mtDNA检测的分析结果显示群体间差异不显著,与RAPD分析结果相悖。对此,作者把两种标记分析结果产生的矛盾归结为两种标记的性质差异,即RAPD检测双亲遗传的核DNA,而mtDNA检测单亲遗传的线粒体DNA。Dumolin-Lapègue 等[50]在该方面提出自己的看法,认为RAPD标记主要从总体上揭示物种的遗传多样性,而母系遗传的线粒体DNA标记更适于描述群体变异和演化路径。
Scotti等[51]应用 mtVNTRs和 cpSSRs标记对小范围内的亚高山挪威云杉群体空间结构和风媒(花粉/种子)更新过程进行研究,该研究所用的材料根据林木胸径大小分为两类,即胸径不小于10 cm的成熟林和胸径小于10 cm的幼龄林,通过cpSSRs标记检测到的单体型多样性(以单体型计数形式表示,nA=42)高于通过mtVNTRs标记检测到的单体型多样性(nA=8),说明叶绿体标记比线粒体标记检测的遗传多样性高。这与之前Nei[52]用相同材料所得结论一致。Nei的研究是应用两种标记对成熟林和幼龄林群体分别单独检测遗传多样性,并将成熟林和幼龄林作为一个整体进行检测,结果表明cpSSRs检测到的遗传多样性0.736高于mtVNTRs检测到的遗传多样性0.478。Scotti将这一现象归因于3点:(i)两种基因组位点的等位基因突变能力存在差异(ii)线粒体和叶绿体基因的遗传属性不同。对于云杉而言,线粒体基因为母系遗传,靠种子传播,具有有限的基因流;叶绿体基因为父系遗传,靠种子和花粉传播,具有广泛的基因流,因此,用后者检测的遗传多样性相对较高。(iii)用于比较的两个基因组的标记数量不同(4个叶绿体标记和2个线粒体标记)。
3 DNA标记在云杉种质资源保存、遗传改良方面的应用前景
在林木育种工作中,对种质资源的保存通常是对遗传多样性资源的保存,即对基因资源的保存。因此,对于云杉而言,通过DNA分子标记研究其天然群体的遗传结构,所获数据可为育种策略的异地保存提供指导。根据现有资料显示,在云杉种质资源保存研究领域,国内外育种专家均主张重视核心种质的保存,以尽可能少的样本尽可能多的保存其遗传多样性,提高种质资源的保存效率。
种质资源的收集与保存是进行遗传改良的遗传基础。云杉属树种早期的遗传改良程序通常是对种和种源进行联合选择,了解不同云杉异地适应性和生产力,再在种源研究基础上进行种子区划,为各造林区提供最适种源。在优良种源区内,还可以进一步选择优良林分,将其改建成母树林,或通过表型选优,营建实生和无性系初级种子园对优良种源进行利用。尽管采用上述方法可达到遗传改良目的,但选育周期很长,而通过分子标记辅助选择育种不仅可以克服上述缺点,还可以同时选择多个性状,是未来云杉遗传育种工作中的一个发展方向。综上,DNA分子标记在云杉属树种的种质资源保存与利用方面具有广阔的前景。
[1]傅淑霞,王文采,郑万钧,等.中国植物志,第7卷[M].北京:科学出版社,1978:123~167.
[2]罗建勋.云杉天然群体遗传多样性研究[D].北京:中国林业科学研究院,2004.
[3]张含国.红皮云杉遗传多样性的研究[D].哈尔滨:东北林业大学,2000.
[4]王芋华.粗枝云杉(Picea asperata Mast.)天然群体的遗传变异[D].成都:中国林业科学研究院(成都生物研究所),2004.
[5]Maghuly F,Burg K,Pinsker W,et al.Short Note:Development of Mitochondrial Markers for Population Genetics of Norway Spruce[Picea abies(L.)Karst][J].Silvae Genetica,2008,57(1):41 ~44.
[6]Murray B G,Davies B J.An improved method for preparing the chromosomes of Pines and other gymnosperms[J].Biotech Histochem,1996,3:115 ~117.
[7]Dering M,Lewandowski A.Finding the meeting zone:Where have the northern and southern ranges of Norway spruce overlapped?[J].Forest Ecology and Management,2009,259:229 ~235.
[8]Bastien D,Favre J M,Collignon A M,et al.Characterization of a mosaic minisatellite locus in the mitochondrial DNA of Norway spruce[Picea abies(L.)Karst.][J].Theor Appl Genet,2003,107:574~580.
[9]Collignon A M,Sype H Van de,Favre J M.Geographical variation in random amplified polymorphic DNA and quantitative traits in Norway spruce[J].Can.J.For.Res.,2002,32:266 ~282.
[10]Jeandroz S,Collignon A M,Favre J M.RAPD and mtDNA variation among autochthonous and planted populations of Picea abies from the Vosges mountains(France)in reference to other French populations[J].Forest Ecology and Management,2004,197:225~229.
[11]Scotti I,Vendramin G G,Matteotti L S.Postglacial recolonization routes for Picea abies K.in Italy as suggested by the analysis of sequence-characterized amplified region(SCAR)markers[J].Molecular Ecology,2000,9:699 ~708.
[12]Wang X R,Chhatre V E,Nilsson M C,et al.Island Population Structure of Norway Spruce(Picea abies)in Northern Sweden[J].International Journal of Plant Sciences,2003,164(5):711~717.
[13]Hodgetts R B,Aleksiuk M A,Brown A.Development of microsatellite markers for white spruce(Picea glauca)and related species[J].Theoretical and Applied Genetics,2001,102:1252 ~1258.
[14]A'Hara S W,Cottrell J E.Development of a set of highly polymorphic genomic microsatellites(gSSRs)in Sitka spruce(Picea sitchensis(Bong.)Carr.)[J].Mol Breeding,2009,23:349 ~355.
[15]Scotti I,Burelli A,Cattonaro F,et al.Analysis of the distribution of marker classes in a genetic linkage map:a case study in Norway spruce(Picea abies)[J].Tree Genetics and Genomes,2005,1:93~102.
[16]Nasri P N,Bojovic S,Vendramin G G,et al.Population genetic structure of the relict Serbian spruce,Picea omorika,inferred from plastid DNA[J].Pl Syst Evol,2008,271:1 ~7.
[17]Ballian D,Longauer R,Mikic T,et al.Genetic structure of a rare European conifer,Serbian spruce(Picea omorika [Panc ˇic']Purkyne ˇ).Pl Syst Evol,260:53 ~ 63.
[18]Anderson L L,Hu F S,Nelson D M,et al.Ice-age endurance:DNA evidence of a white spruce refugium in Alaska[J].PNAS,2006,103(33):12447 ~ 12450.doi:10.1073/pnas.0605310103.
[19]Aizawa M,Yoshimaru H,Saito H,et al.Phylogeography of a northeast Asian spruce,Picea jezoensis,inferred from genetic variation observed in organelle DNA markers[J].Molecular Ecology,2007:3393 ~3405.
[20]Zietkiewicz E,Rafalski A,Labuda D.Genomic fingerprinting by simple sequence repeat(SSR)-anchored polymerase chain reaction amplification[J].Genomics,1994,20:176 ~183.
[21]Dobrzeniecka S,Nkongolo K K,Michael P,et al.Genetic Analysis of Black Spruce(Picea mariana)Populations from Dry and Wet Areas of a Metal-Contaminated Region in Ontario(Canada)[J].Water Air Soil Pollut,2011,215:117 ~125.
[22]Perry J,Bousquet J.Genetic diversity and mating system of postfire and post-harvest black spruce:an investigation using codominant sequence-tagged-site(STS)markers[J].Canadian Journal of Forest Research,2011,31:32 ~41.
[23]Rajora O P,Pluhar S A.Genetic diversity impact of forest fires,forest harvesting and alternative reforestation practices in black spruce(Picea mariana)[J].Theoretical and Applied Genetics,2003,106:1203 ~1212.
[24]Cardle L,Ramsay L,Milbourne D,et al.Computational and experimental characterization of physically clustered simple sequence repeats in plants[J].Genetics,2000,156:847 ~ 854.
[25]Tóth G,Gáspári Z,Jurka J.Microsatellites in different eukaryotic genomes:survey and analysis[J].Genome Res,2000,10:967 ~981.
[26]Temnykh S,De Clerck G,Lukashova A,et al.Computational and experimental analysis of microsatellites in rice(Oryza sativa L.):frequency,length variation,transposon associations and genetic marker potential[J].Genome Res,2001,11:1441 ~ 1452.
[27]Kantety R V,La Rota M,Matthews D E,et al.Data mining for simple sequence repeats in expressed sequence tags from barley,maize,rice,sorghum and wheat[J].Plant Mol Biol,2002,48:501~510.
[28]Varshney R K,Thiel T,Stein N,et al.In silico analysis on frequency and distribution of microsatellites in ESTs of some cereal species[J].Cell Mol Biol Lett,2002,7:537 ~ 546.
[29]Gao L,Tang J,Li H,et al.Analysis of microsatellites in major crops assessed by computational and experimental approaches[J].Mol Breed,2003,12:245 ~261.
[30]Fluch S,Burg A,Kopecky D,et al.Characterization of variable EST-SSR markers for Norway spruce(Picea abies L.)[Online].BMC Research Notes,2011,4:401.[http://www.biomedcentral.com/content/pdf/1756-0500-4-401.pdf]
[31]Bérubé Y,Zhuang J,Rungis D,et al.Characterization of ESTSSRs in loblolly pine and spruce[J].Tree Genetics and Genomes,2007,3:251 ~259.
[32]A’Hara S W,Cottrell J E.Characterization of a suite of 40 EST-derived microsatellite markers for use in Sitka Spruce(Picea sitchensis(Bong.)Carr)[J].Silv.Gen.,2007,56:138 ~141.
[33]Rungis D,Berube Y,Zhang J,et al.Robust simple sequence repeat markers for spruce(Picea spp.)from expressed sequence tags[J].Theor.Appl.Genet.,2004,109:1283 ~1294.
[34]Olson M,Hood L,Cantor C,et al.A common Language for physical mapping of the human genome[J].Science,1989,245:1434~1435.
[35]Bennuah S Y,Wang T L,Aitken S N,et al.Genetic analysis of the Picea sitchensis×glauca introgression zone in British Columbia[J].Forest Ecology and Management,2004,197:65 ~ 77.
[36]Gapare W J,Aitken S N,Ritland C E.Genetic diversity of core and peripheral Sitka spruce(Picea sitchensis(Bong.)Carr)populations:implications for conservation of widespread species[J].Biological Conservation,2005,123:113 ~123.
[37]Gapare W J.Genetic Diversity and Spatial Population Structure of Sitka Spruce(Picea sitchensis(Bong.)Carr.):Implications for Gene Conservation of Widespread Species[D].University of British Columbia,2003:148.
[38]Holliday J A,Ralph S G,White R,et al.Global monitoring of autumn gene expression within and among phenotypically divergent populations of Sitka spruce(Picea sitchensis)[J].2008,New Phytologist,178:103 ~ 122.
[39]Perkel J.SNP genotyping:Six technologies that keyed a revolution[J].Nature Methods ,2008,5:447 ~453.
[40]Namroud M C,Beaulieu J,Juge N,et al.Scanning the genome for gene single nucleotide polymorphisms involved in adaptive population differentiation in white spruce[J].Molecular Ecology,2008,17(16):3599 ~3613.
[41]Holliday J A,Ritland K,Aitken S N.Widespread,ecologically relevant genetic markers developed from association mapping of climate-related traits in Sitka spruce(Picea sitchensis)[J].New Phytologist,2010,188:501 ~ 514.
[42]Ayala F J,Kiger J A.Modern Genetics(2nd)[M].Benjamin:Cummings Publishing Company,1984.
[43]Hamrick J L,Godt M L.Allozyme Diversity in Plants Species[A].In:Brown A H D,Clegg M T,Kahler A L,et al.(eds),Plant Population Genetics,Breeding and Germplasm Resources[M].Sunderland:Sinauer Associates,Inc,1989,43 ~63.
[44]Qian W,Ge S,Hong D Y.Genetic variation within and among populations of a wild rice Oryza granulata from China detected by RAPD and ISSR markers[J].Theor Appl Genet,2001,102:440~449.
[45]Collignon A M,Favre J M.Contribution to the postglacial history at the western margin of Picea abies’natural area using RAPD markers[J].Ann Bot,2000,85:713 ~ 722.
[46]Dobrzeniecka S,Nkongolo K K,Michael P,et al.Genetic Analysis of Black Spruce(Picea mariana)Populations from Dry and Wet Areas of a Metal-Contaminated Region in Ontario(Canada)[J].Water Air Soil Pollut,2011,215:117 ~125.
[47]Maghuly F,Burg K,Pinsker W,et al.Short Note:Development of Mitochondrial Markers for Population Genetics of Norway Spruce[Picea abies(L.)Karst][J].Silvae Genetica,2008,57(1):41~44.
[48]Namroud M C,Beaulieu J,Juge N,et al.Scanning the genome for gene single nucleotide polymorphisms involved in adaptive population differentiation in white spruce[J].Molecular Ecology ,2008,17(16):3599 ~3613.
[49]Jeandroz S,Collignon A M,Favre J M.RAPD and mtDNA variation among autochthonous and planted populations of Picea abies from the Vosges mountains(France)in reference to other French populations[J].Forest Ecology and Management,2004,197:225~229.
[50]Dumolin-Lapègue S,Demesure B,Fineschi S,et al.Phylogeographic structure of white oaks throughout the European continent[J].Genetics,1997,146:1475 ~ 1487.
[51]Scotti I,Gugerli F,Pastorelli R,et al.Maternally and paternally inherited molecular markers elucidate population patterns and inferred dispersal processes on a small scale within a subalpine stand of Norway spruce(Picea abies[L.]Karst.)[J].Forest Ecology and Management,2008,255:3806 ~3812.
[52]Nei M.Analysis of gene diversity in subdivided populations[J].Proceedings of the National Academy of Sciences of the United States of America,1973,70:3321 ~33.
[53]Meloni M,Perini D,Binelli G.The distribution of genetic variation in Norway spruce(Picea abies Karst.)populations in the western Alps[J].Journal of Biogeography,2007,34(6):929 ~938.
