氧分子在黄铁矿和方铅矿表面的吸附
2012-11-24李玉琼陈建华蓝丽红
李玉琼,陈建华,蓝丽红,,郭 进
(1. 广西大学 化学化工学院,南宁 530004;2. 广西大学 资源与冶金学院,南宁 530004;3. 广西民族大学 化学与生态工程学院,南宁 530006;4. 广西大学 物理科学与工程技术学院,南宁 530004)
氧分子在黄铁矿和方铅矿表面的吸附
李玉琼1,陈建华2,蓝丽红1,3,郭 进4
(1. 广西大学 化学化工学院,南宁 530004;2. 广西大学 资源与冶金学院,南宁 530004;3. 广西民族大学 化学与生态工程学院,南宁 530006;4. 广西大学 物理科学与工程技术学院,南宁 530004)
采用密度泛函理论对氧分子在黄铁矿和方铅矿表面的吸附进行研究。计算结果表明:黄铁矿和方铅矿表面经历了较小的弛豫;氧分子在黄铁矿和方铅矿表面都呈解离吸附状态,且在黄铁矿表面的吸附能远低于在方铅矿表面的吸附能;在黄铁矿表面上,氧原子分别与铁原子和硫原子键合,电子由铁原子和硫原子转移到氧原子上,主要由硫的3p态、氧的2p态和铁的3d态参与反应,铁与氧之间形成d→p反馈键,而在方铅矿表面上,氧原子只与硫原子键合,主要由硫的3p态、氧的2p态和铅的6p态参与反应,未形成反馈键;氧吸附后黄铁矿表面产生键合的铁原子和氧原子都产生自旋现象,而方铅矿表面原子及吸附的氧原子仍然是低自旋态的。
黄铁矿;方铅矿;氧分子吸附;密度泛函理论
有色金属硫化矿浮选是一个电化学过程,矿物表面的氧化对其浮选行为具有决定性的影响,硫化矿物的浮选行为与表面氧化之间存在密切的关系[1-3]。已有研究证实:捕收剂黄药在硫化矿物表面的吸附是一个电化学反应过程,黄药在硫化矿表面发生阳极氧化生成金属黄原酸盐或双黄药,氧分子在矿物表面发生阴极还原。氧分子在硫化矿表面吸附对黄药捕收的影响非常大。另外,硫化矿无捕收剂浮选主要是通过氧化来控制矿物表面产物形成疏水元素硫和亲水硫酸盐,实现矿物的浮选和抑制[4-5]。因而氧分子在硫化矿浮选中的作用成为硫化矿浮选研究中最重要的理论问题之一。
黄铁矿和方铅矿是浮选电化学过程中最具代表性的两种硫化矿,捕收剂黄药分别在黄铁矿表面和方铅矿表面形成疏水的双黄药和金属黄原酸盐,代表硫化矿捕收作用的两种典型电化学机制,另一方面,方铅矿具有良好的无捕收剂浮选行为,而黄铁矿则较差。不论是黄药捕收剂浮选还是无捕收剂浮选,都与氧分子在这两种矿物表面的还原密不可分。目前的研究多集中在硫化矿表面氧化动力学和表面化学方面[6-15]。RAIKAR等[16]采用自然黄铁矿作为研究对象,发现将黄铁矿暴露在氧气中铁组分将出现严重的氧化现象,而ROSSO等[17]也发现真空解理的黄铁矿与氧作用后表面形成了Fe—O键,此外,KNDELEWICZ等[18]则发现,黄铁矿仅暴露给水没有导致硫组分氧化,而单独暴露给氧则导致了硫组分的氧化。而对于氧分子在黄铁矿和方铅矿表面上的吸附方式、吸附构型以及矿物表面电子转移情况等详细的吸附机理却缺少研究。王淀佐等[19]采用分子轨道法针对黄铁矿和方铅矿的氧化进行了研究,但他们只采用单层原子来模拟矿物表面,因此,其计算结果很难与实际表面吸附相结果一致。ROSSO等[17]采用试验和丛簇计算相结合的方法,表明氧在黄铁矿表面解离吸附。SUN等[20]采用密度泛函方法简单研究了氧分子在黄铁矿表面的吸附,但没有考虑黄铁矿和氧分子的自旋。
本文作者通过构造含多层原子的矿物表面层晶模型,采用密度泛函理论对黄铁矿和方铅矿表面弛豫和氧分子在表面上的吸附方式、吸附能、表面电荷分布和电子转移及表面态进行详细的研究,研究结果对进一步阐明氧气在硫化矿电化学浮选中的作用具有重要意义。
1 计算方法和模型
1.1 计算方法
本文作者采用基于密度泛函理论和平面波赝势方法的CASTEP软件[21-22]进行计算。交换关联泛函采用广义梯度近似(GGA)下的PW91梯度修正近似。平面波截断能经过测试后对黄铁矿选取270 eV,对方铅矿选取280 eV。表面由优化过的体相切出,并在吸附前进行优化,为了消除相邻吸附分子之间的相互影响,分别对黄铁矿和方铅矿采用(2×2)和(4×2)表面层晶模型。进行氧分子吸附计算时,将氧分子置于不同的位置上,如垂直置于硫位、铅位、铁位和穴位以及平躺等方式,通过优化构型、计算吸附能,确定最稳定的吸附模式。对价电子和离子实的相互作用势的描述采用超软赝势(USP)[23],Brillouin区的积分计算分别对黄铁矿采用 2×2×1和对方铅矿采用 1×2×1的Monkhorst-Pack(MP)k点网络[24-25]。计算的价电子构型为Fe 3d64S2,S 3s23p4和Pb 6s26p2。几何优化采用BFGS算法,对黄铁矿进行优化的收敛标准如下:能量收敛标准为 2.0×10-5eV·atom-1,原子位移的收敛标准为0.000 2 nm,原子间作用力的收敛标准为0.8 eV·nm-1,晶体内应力收敛标准为0.1 GPa;自洽迭代收敛精度为 2.0×10-6eV·atom-1;除对方铅矿进行优化的收敛标准除原子间作用力的收敛标准采用 0.5 eV·nm-1外,其余收敛标准与黄铁矿相同。对含不同原子层及真空层厚度的表面进行了计算,以获得较稳定的表面,根据最终的测试结果确定:对于黄铁矿,采用15层原子层以及15 Å埃真空层厚度的表面模型(考虑到需要吸附氧分子,所以真空层取值大一点),并在吸附过程中固定基底9层原子层,对表面6层原子进行弛豫;对于方铅矿,采用8层原子层,真空层厚度的采取与黄铁矿一致,吸附过程中固定基底5层原子层,对表面3层原子进行弛豫;对于氧分子,则将其放入一个1.5 nm×1.5 nm×1.5 nm的盒子中,计算时采用Gamma点。所有计算都采用自旋极化方法。
1.2 计算模型
1.3 吸附能计算
氧分子在黄铁矿和方铅矿表面的吸附能按下式定义:
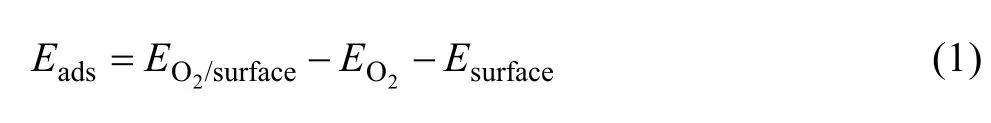
其中:Eads为氧分子吸附后的吸附能; EO2/surface为氧分子在表面吸附后体系的总能; EO2和 Esurface分别为吸附前氧分子和表面的总能。吸附能越低,吸附越稳定,反之则稳定性降低。
2 结果与讨论
2.1 黄铁矿和方铅矿表面结构弛豫
计算得到黄铁矿和方铅矿的晶格常数分别为0.542 1和0.601 8 nm,分别与实验值0.541 7和0.593 6 nm非常接近[26-27],优化后的氧分子中 O—O键长为0.124 1 nm,也与实验值0.120 9 nm非常接近,表明计算是可靠的。表1和表2所列分别为弛豫后黄铁矿和方铅矿表面几层原子的配位数及位移,其中负号表明原子沿轴的负方向弛豫,反之则沿轴正方向弛豫。在黄铁矿表面,表面第一层S原子向表面内部弛豫;最明显的弛豫是第二层的表面铁原子,向内部弛豫约0.01 nm;第三层中的S原子则向表面弛豫。原子仅在顶部3层产生了明显的弛豫,第四层至第六层原子经历了微小的位移,第七层至第九层原子的弛豫可以忽略不计。这与 ROSSO等[28]及 CHATURVEDI等[29]的实验测试结果一致,也与 HUNG 等[30]的计算结果一致。对于方铅矿表面,第一层的硫原子和铅原子向表面内部弛豫,而第二层的原子都沿z轴方向表面外部弛豫,且这一层的硫原子和铅原子弛豫最为明显,第三层原子都向表面内部弛豫,且弛豫较小。
表面弛豫计算表明,黄铁矿和方铅矿表面解理后都发生了不同程度的表面弛豫,但没有产生明显的表面重构作用,且仅有顶部3层原子的弛豫略微明显,更低层原子的弛豫非常小。
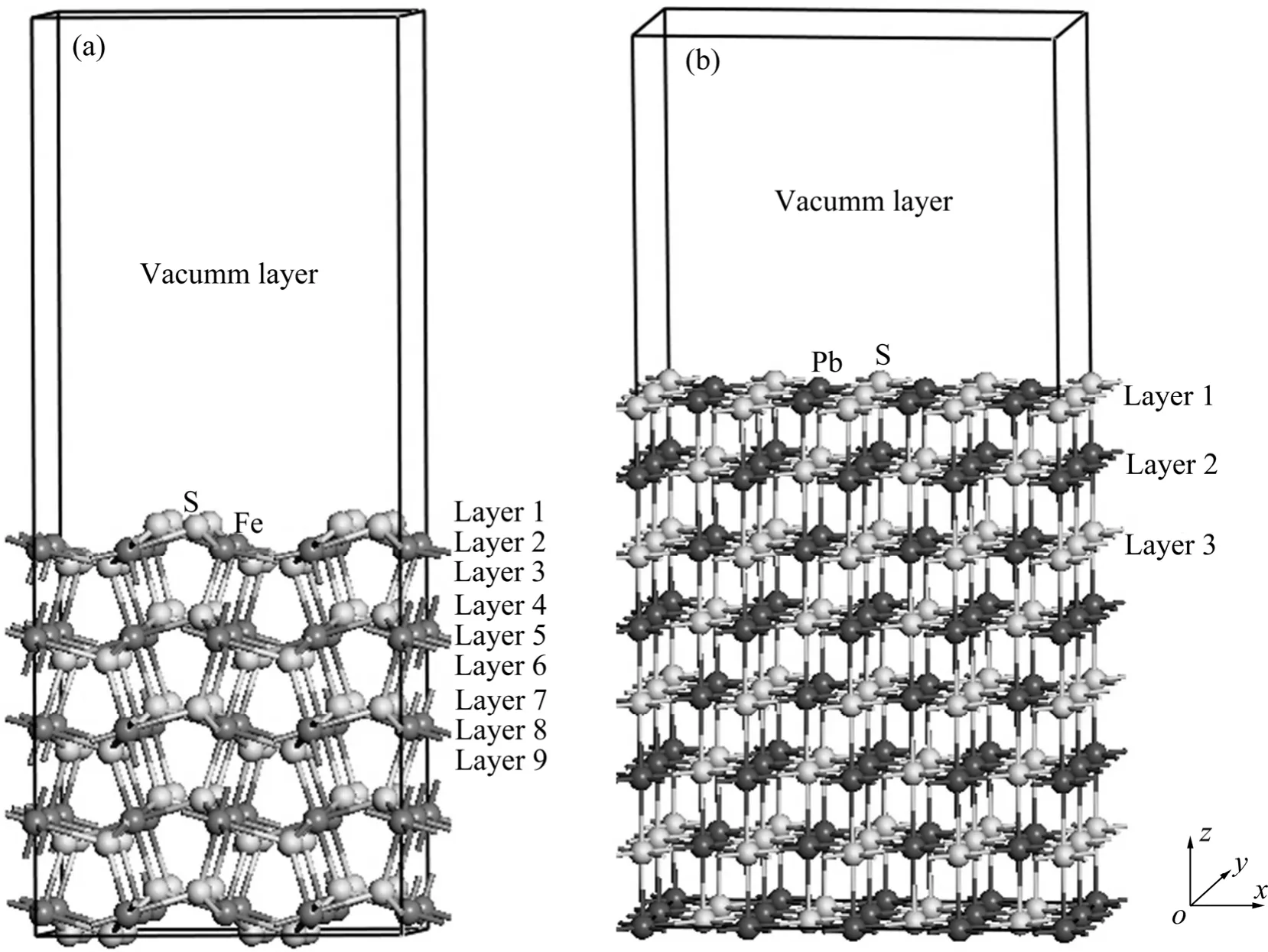
图1 (2×2)黄铁矿(100)表面层晶模型以及(4×2)方铅矿(100)表面层晶模型Fig. 1 Slab models of (2×2) pyrite (100) surface (a) and (4×2) galena (100) surface (b)

表1 黄铁矿表面原子配位及位移Table 1 Atomic coordination and displacements of pyrite surface
2.2 氧分子在黄铁矿和方铅矿表面的吸附
为了确定氧分子在黄铁矿和方铅矿表面的吸附方式,分别对氧分子在各个吸附位进行了测试,结果如图2和图3所示。并计算了吸附能,计算结果如表3和表4所列。吸附能计算结果表明,在黄铁矿表面,氧分子在顶部硫位(见图 2(a))、平行于 S—S键(见图2(d))、垂直于穴位(见图2(e))吸附时的吸附能较高,而在顶部铁位(见图2(b))、平行于Fe—S键(见图2(c))和平躺在穴位(见图2(f))吸附时的吸附能较低,且以平躺在穴位上以一个氧原子对着顶部硫原子、另一个氧原子对着表面铁原子(见图 2(g))时的吸附能最低,表明这种吸附方式最为稳定;在方铅矿表面,除氧分子以平躺于穴位且两个氧原子分别对着两个硫原子(见图3(f))吸附在表面时的吸附能最低,吸附最稳定外,其他吸附方式:顶部硫位(见图 3(a))、顶部铅位(见图3(b))、平行于S—Pb键(见图3(c))、平躺在穴位(处于铅原子之间)(见图 3(d))以及垂直于穴位(见图 3(e))的氧分子吸附能都较高。

表2 方铅矿表面原子配位及位移Table 2 Atomic coordination and displacements of galena surface

图2 氧分子在黄铁矿(100)面不同位置的平衡吸附构型Fig. 2 Equilibrium adsorption of O2 on different sites of pyrite (100) surface (Numbers shown near bond indicating bond length in nm. Arrows are indicators of x, y and z axes): (a) Top S site; (b) Top Fe site; (c) Parallel to Fe—S bond; (d) Parallel to S—S bond;(e) Vertical to hollow; (f) Parallel to hollow (between Fe atoms); (g) Parallel to hollow (between Fe and S atoms)
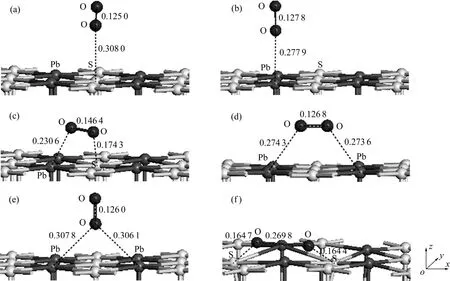
图3 氧分子在方铅矿(100)面不同位置的平衡吸附构型Fig. 3 Equilibrium adsorption of O2 on different sites of galena (100) surface (Numbers shown near bond indicating bond length in nm. Arrows are indicators of x, y and z axes): (a) Top S site; (b) Top Pb site; (c) Parallel to Pb—S bond; (d) Parallel to hollow(between Pb atoms); (e) Vertical to hollow; (f) Parallel to hollow (between S atoms)

表3 氧分子在黄铁矿(100)面的吸附能Table 3 Adsorption energy of O2 on pyrite (100) surface

表4 氧分子在方铅矿(100)面吸附时的吸附能Table 4 Adsorption energy of O2 on galena (100) surface
在黄铁矿和方铅矿表面吸附后的氧分子都发生了解离,并分别与表面的原子成键。从氧分子在两种矿物表面上的最稳定吸附方式可以知道,在黄铁矿表面,氧原子分别与硫和铁原子键合,而在方铅矿表面,氧原子只与硫原子键合而未与铅原子键合。氧分子在黄铁矿和方铅矿表面的吸附能分别为-2.522 eV(见图2(g))和-1.191 eV(见图 3(f)),前者明显低于后者,表明其与黄铁矿表面的相互作用更强,在黄铁矿表面的反应活性更高,这也体现在不同表面吸附后的 O—O键长和O—S键长的区别中。在黄铁矿和方铅矿表面上,O—O键长分别为0.284 2和0.269 8 nm,氧分子在黄铁矿表面的解离更彻底;O—S键长分别为0.149 6和0.164 4 nm,氧原子与黄铁矿表面的硫原子之间的键合更为紧密。从以上的分析可以知道,当氧分子吸附后,黄铁矿表面上的硫原子被氧化得更为彻底,即所带正价将更高,这与实际情况相符,即黄铁矿的阳极氧化产物主要硫组分为硫酸盐(S O),而方铅矿的阳极氧化产物主要硫组分为元素硫(S0)[3]。这也表明黄铁矿具有较差的无捕收剂浮选特性,而方铅矿具有较好的无捕收剂浮选行为。
2.3 表面原子电荷分析
图4所示为黄铁矿和方铅矿表面原子的Mulliken电荷,原子上的数字表示电荷值,单位为 e,图为顶视图。黄铁矿(100)表面硫二聚体中的 S1原子位于表面顶部,S2原子位于表面底部(见图4(a))。从图4(a)可以看出,S1原子带电荷-0.10 e,而S2原子带电荷-0.02 e。另外,与铁原子配位不同方向上的硫原子所带电荷不同,处于表面底部的硫原子(S2和S3)所带负电荷少于表面顶部的硫原子(S4和S5)。表面铁原子带正电荷0.08 e。从氧分子吸附后的表面原子电荷(见图4(b))可以看出,与氧成键的 S1和Fe1原子失去了较多的电荷给氧原子而分别带正电荷0.74 e和0.36 e,而与S1成键的O1原子所带负电荷(-0.76 e)远多于与Fe1原子成键的O2原子(-0.44 e),表明氧原子从表面硫原子上获得的电荷多于从铁原子上获得的电荷。另外,除与表面顶部 S1原子配位的硫原子和其余铁原子(除Fe1)得到少量电荷外,氧分子周围的其余硫原子则失去少量电荷。氧分子吸附对更远处的表面原子的电荷影响较小。
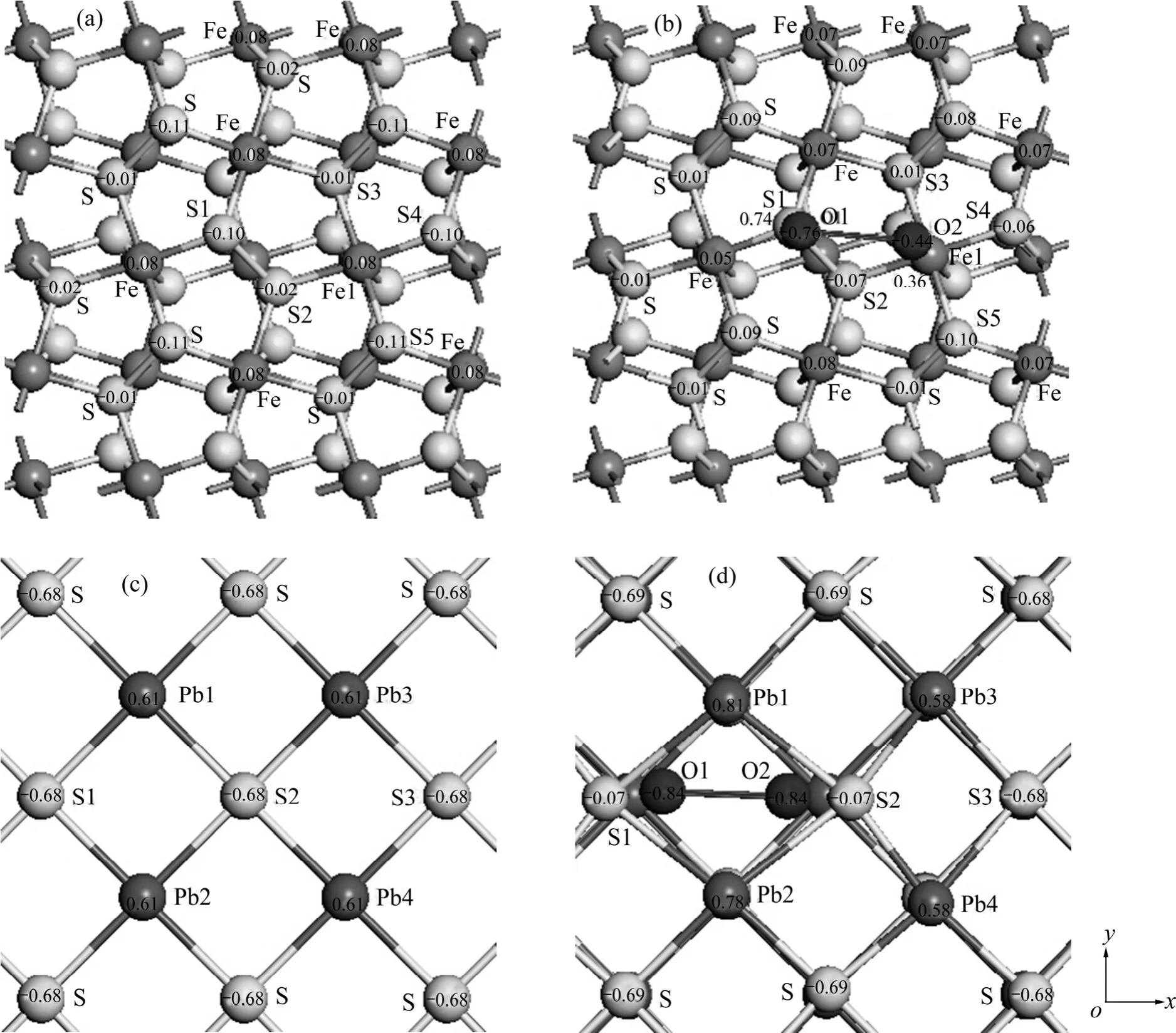
图4 氧分子吸附前后氧原子及表面原子的Mulliken电荷Fig. 4 Mulliken charges of oxygen atom and surface atoms before and after O2 adsorption (Numbers on atom indicating atomic charge in e. Arrows indicating x and y axes): (a) Pyrite surface before O2 adsorption; (b) Pyrite surface after O2 adsorption; (c) Galena surface before O2 adsorption; (d) Galena surface after O2 adsorption
在理想的方铅矿(100)表面上,铅原子带正电荷0.61 e而硫原子带负电荷-0.68 e(见图4(c)),氧分子吸附后对其周围原子的电荷影响较为明显。分别与氧原子成键的S1和S2原子所带电荷已从原来的负电荷到吸附氧后略带正电荷(0.07 e),氧对距离稍远的硫原子电荷影响很小,靠近氧原子的铅原子(Pb1和Pb2)失去电子,而离氧较远的铅原子(Pb3和Pb4)则得到极少量的电子,由原来的0.61 e变为0.58 e。另外,氧分子吸附对表面硫原子的构型产生了较为明显的影响,与氧成键的S1和S2原子沿着x轴被排斥开来。
从更详细的原子电荷布居分析可以了解原子之间的电荷转移情况,表5和表6所列分别为氧分子在黄铁矿和方铅矿表面吸附前和吸附后的表面原子及氧原子的电荷布居值。由表5可以知道,氧分子在黄铁矿表面吸附后,与氧原子成键的 S1原子(顶部硫)的 3s轨道失去少量电子而3p轨道都失去较多电子,离氧稍远处的S2和S3原子的3s轨道电子基本没有变化,但3p轨道失去非常少量电子;铁原子(Fe1)的4s轨道电子不变,4p轨道得到非常少量的电子,而3d轨道则失去了较多电子;氧原子的 2s轨道电子没有发生变化,与S1成键的O1原子的2p轨道比与Fe1原子成键的O2原子的2p轨道得到更多的电子。由此可知,氧分子与黄铁矿表面的反应,主要由硫原子的 3p轨道、铁原子的3d轨道和氧原子的2p轨道参与。
由表6可以看出,氧分子在方铅矿表面吸附后,与氧成键的硫原子(S2)的3s轨道失去少量电子,而3p轨道则失去大量的电子;氧分子周围的铅原子(Pb1和Pb2)的 6s轨道电子基本没有变化,6p轨道失去较多电子;离氧吸附位置稍远的铅原子(Pb3)的 6s轨道电子基本没有变化,而6p轨道则得到少量电子。此外,铅原子的5d轨道电子数没有变化,表明5d轨道电子没有参与氧气的反应。氧的2s轨道得到少量电子,而2p轨道得到较多的电子。由此可知,氧分子与方铅矿表面的反应,主要由硫原子的3p轨道、铅原子的6p轨道和氧原子的2p轨道参与。
从图5的电荷差分密度(见图5(a)和(b),黑色区域表示电子富集,白色区域表示电子缺失,背景色表示电子密度为零)和电荷密度图(见图 5(c)和(d))可以看到,吸附后氧原子周围电子富集而与之配位的铁原子和硫原子周围则呈电子缺失状态,氧与矿物表面的原子发生相互作用而成键,而由于在表面解离成单氧状态,氧原子之间已经不成键。
2.4 氧分子吸附对黄铁矿和方铅矿表面态的影响
从前面的原子电荷布居分析可知:氧分子在黄铁矿和方铅矿表面吸附时,主要是氧原子、硫原子和铅原子的p轨道以及铁原子的d轨道参与相互作用,因此,图6和图7所示分别为氧分子吸附前后黄铁矿和方铅矿表面及表面主要参与反应的原子态密度,并考察参与反应的主要电子轨道态密度变化。氧的外层 p电子组态为:本计算结果与实际非常一致。如图所示,在-6和-4.5 eV处有一个分别由成键σ2pz态和π2px(π2py)组成的态密度峰(px和py的原子轨道态密度曲线是重合的);费米能级处的态密度则由半满的反键)态组成;最后,在约1.8 eV处存在一个空反键态。
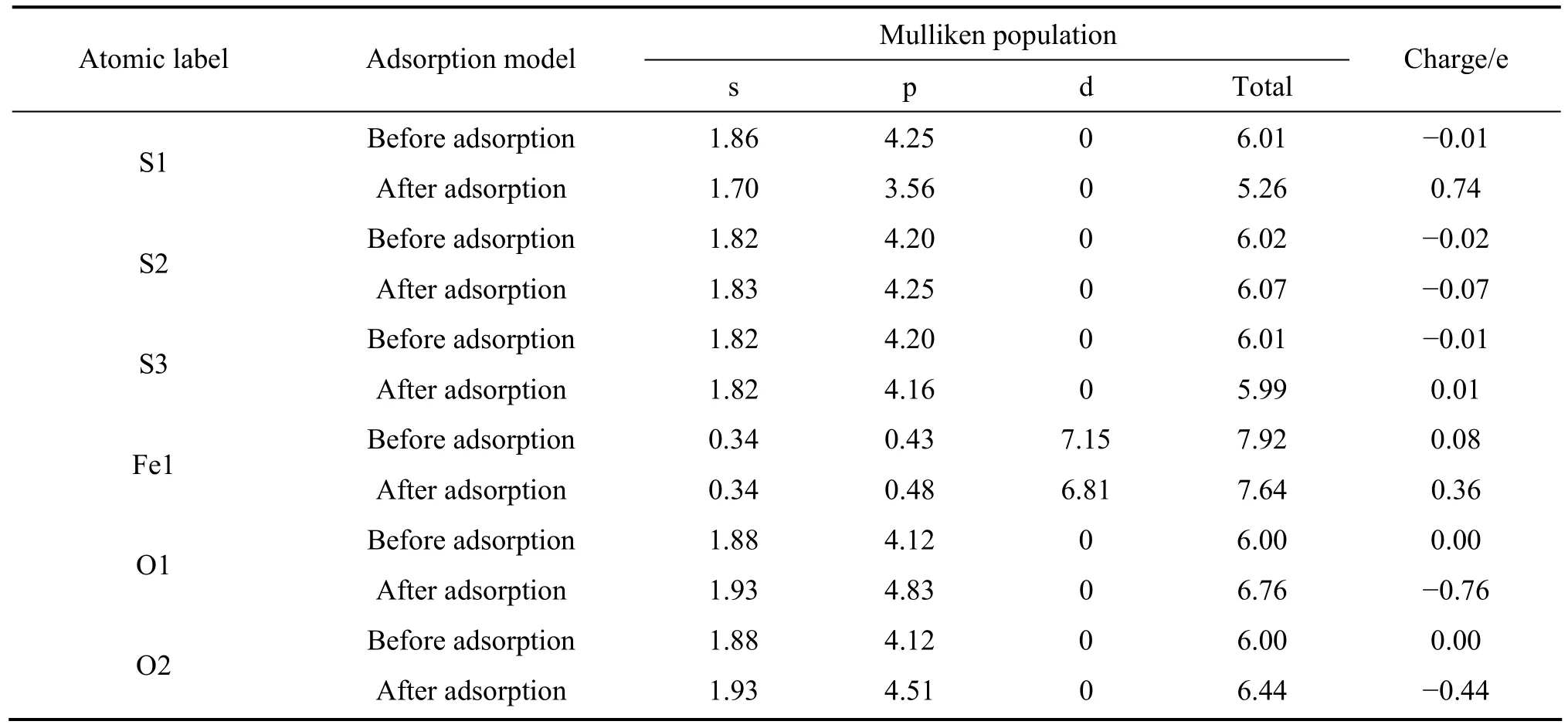
表5 氧分子吸附前后黄铁矿表面原子及氧原子的Mulliken电荷布居Table 5 Mulliken charge populations of O atom and surface atom before and after O2 adsorption on pyrite surface

表6 氧分子吸附前后方铅矿表面原子及氧原子的Mulliken电荷布居Table 6 Mulliken charge populations of O atom and surface atom before and after O2 adsorption on galena surface

图5 氧分子吸附后黄铁矿和方铅矿的电荷密度和差分电荷密度Fig. 5 Electron density and electron density difference maps of pyrite and galena after O2 adsorption: (a) Electron density difference of pyrite; (b) Electron density difference of galena; (c) electron density of pyrite; (d) Electron density of galena
在黄铁矿表面,与氧成键的硫原子和铁原子的态密度发生了极为明显的变化,而氧气分子本身的态密度也发生了变化,说明氧吸附对矿物表面态产生了明显的影响。本文作者主要通过讨论占据电子态来讨论氧与表面的相互作用。费米能级以下-8~0 eV能量范围内吸附氧的2p轨道电子态密度呈连续分布状态,电子非局域性增强;吸附氧后的 S1原子在费米能级以下-8~-1.50 eV范围内的3p电子态密度峰向低能方向移动,而-1.5~0 eV的3p电子态密度明显降低;Fe1原子的3d轨道电子对态密度的贡献占主导地位,吸附氧后在-6~0 eV范围内形成连续分布,并且-1.5~0 eV范围内的 3d电子态密度明显降低。氧吸附对方铅矿表面也产生了明显影响。在费米能级以下-6~0 eV范围内吸附后的氧2p轨道电子态密度呈连续分布状态,且主要集中在-5.5~3.5 eV能量范围内;S2原子由于吸附氧导致原来处于-4~0 eV范围的连续3p电子态在-6~0 eV 能量范围内形成两个集中态,即集中在-5~-4 eV和-1~0 eV能量范围内;Pb1原子的整体态密度向低能方向移动了较小距离且 6p电子态密度明显降低(5d轨道电子能量非常低,在氧吸附过程中没有参与反应),-3~-1 eV能量范围内的6p电子态由于氧吸附而几乎消失。
由态密度图可以看出,在黄铁矿表面,氧与硫成键时,电子主要由硫的3p轨道向氧的2p轨道转移,而与铁成键时,则主要由铁的3d轨道电子向氧的2p轨道转移,形成d→p反馈键;在方铅矿表面上,电子主要由硫和铅的6p轨道向氧的2p轨道转移,而铅的5d轨道由于没有参与反应,未能与氧的2p轨道形成d→p反馈键。因氧与铁之间d→p反馈键的形成,氧分子在黄铁矿表面的吸附将更为稳定,黄铁矿表面将被氧化得更为彻底,这与前面对吸附能和键长的计算的结果一致。

图6 氧分子吸附前后黄铁矿表面原子氧气态密度Fig. 6 Density of states of surface atom and oxygen before and after O2 adsorption on pyrite surface (EF indicates position of Fermi level, and the value is 0 eV)
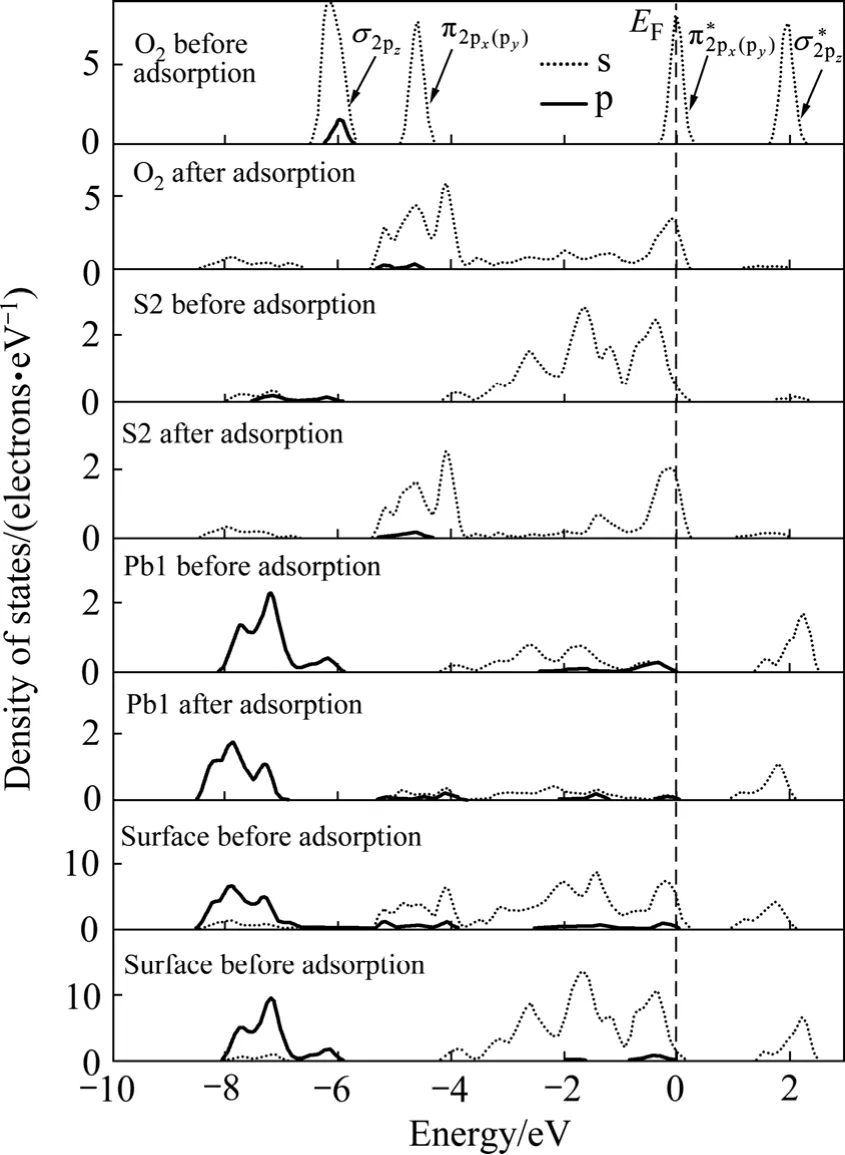
图7 氧分子吸附前后方铅矿表面原子和氧气态密度Fig. 7 Density of states of surface atom and oxygen before and after O2 adsorption on galena surface

图8 氧分子吸附前后黄铁矿表面原子和氧原子的自旋态密度Fig. 8 Spin density of states of surface atom and oxygen atom before and after O2 adsorption on pyrite surface
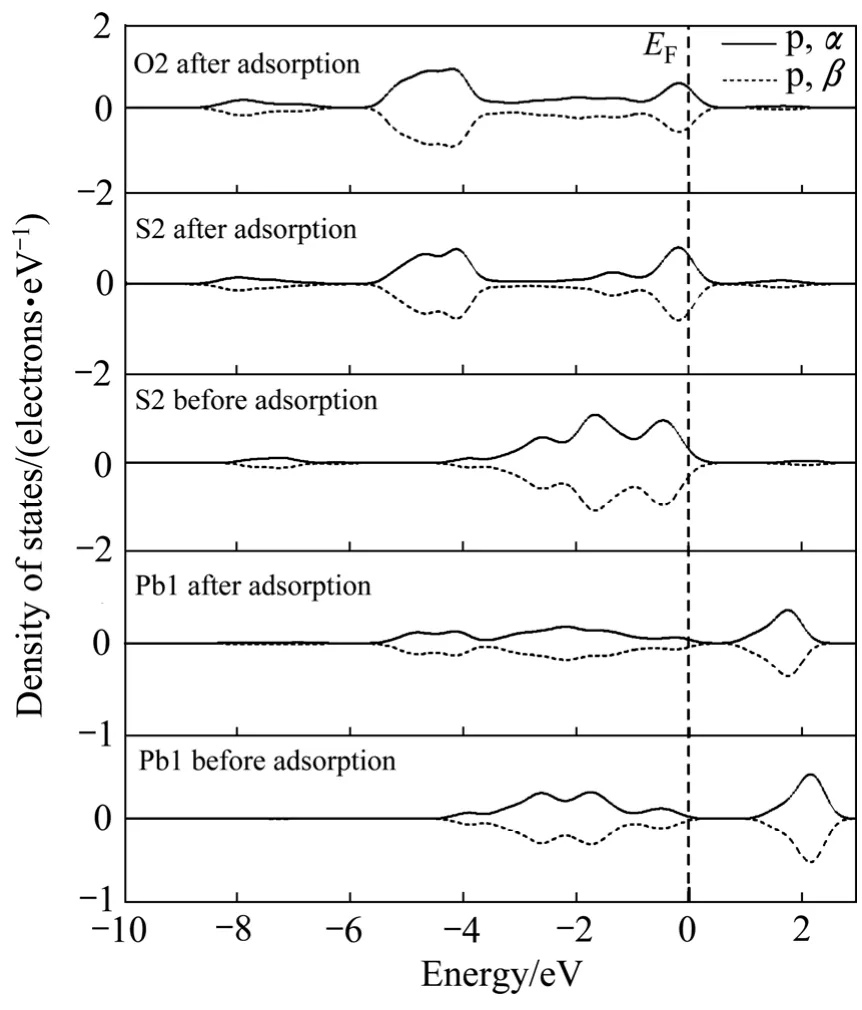
图9 氧分子吸附前后方铅矿表面原子和氧原子的自旋态密度Fig. 9 Spin density of states of surface atom and oxygen atom before and after O2 adsorption on galena surface
图8和9所示分别为氧分子吸附前后的黄铁矿和方铅矿表面原子自旋态密度。图中仅显示了主要参与反应的轨道电子自旋,即S 3p、O 2p、Fe 3d和Pb 6p轨道电子,主要考察其在费米能级(EF)附近的变化,α和β分别代表向上和向下自旋。由图8可以看出,氧分子吸附前的铁原子(Fe1)为低自旋态,吸附后产生了自旋,而与铁原子成键的氧原子(O2)也产生了自旋;吸附前后的硫原子(S1)都呈低自旋态,与硫原子成键的氧原子(O1)也为低自旋态。由图9可以看出,吸附前后氧、硫和铅原子都呈低自旋态,没有产生自旋现象。从电子自旋分析可以看出,在黄铁矿表面上的氧和铁原子由于发生相互作用而产生了自旋现象,而在方铅矿表面的氧则没有发生自旋现象,具有磁性的物质之间更容易产生相互吸引,因此,氧分子在黄铁矿表面的反应活性将比在方铅矿表面大。
3 结论
1) 氧分子在黄铁矿和方铅矿表面具有明显不同的吸附方式。氧分子和黄铁矿作用产生了自旋,导致氧分子在黄铁矿表面的反应活性更高,而氧分子与方铅矿表面相互作用后没有产生自旋,削弱了氧分子在方铅矿表面的吸附。
2) 在黄铁矿表面,氧与金属铁原子之间形成d→p反馈键,而在方铅矿表面没有反馈键形成,这也增强了氧在黄铁矿表面的吸附;黄铁矿表面氧化倾向于生成高价硫,氧化更为彻底,而方铅矿表面氧化后倾向于形成低价硫,这与实际检测结果一致,从理论上解释了黄铁矿和方铅矿无捕收剂浮选差异的本质原因。
REFERENCES
[1] PLASKIN I N. Interaction of minerals with gases and reagents in flotation[J]. Mining Engineering, 1959, 214: 319-324.
[2] HOFFMAN I, NATHANIEL A. Flotation[J]. Industrial and Engineering Chemistry, 1957, 49(3): 493-496.
[3] 冯其明, 陈 荩. 硫化矿物浮选电化学[M]. 长沙: 中南工业大学出版社, 1992: 14-90.FENG Qi-ming, CHEN Jin. Electrochemistry of sulfide mineral flotation[M]. Changsha: Central South University of Technology Press, 1992: 14-90.
[4] WOODS R. Electrochemical potential controlling flotation[J].International Journal of Mineral Processing, 2003, 72(1/4):151-162.
[5] WOODS R. Recent advances in electrochemistry of sulfide mineral flotation[J]. Transactions of Nonferrous Metals Society of China, 2000, 10(Special Issue): 26-29.
[6] UHLIG I, SZARGAN R, NESBITT H W, LAAJALEHTO K.Surface states and reactivity of pyrite and marcasite[J]. Applied Surface Science, 2001, 179(1/4): 222-229.
[7] TOSSELL J A, VAUGHAN D J. Electronic structure and the chemical reactivity of the surface of galena[J]. Canadian Mineralogist, 1987, 25: 381-392.
[8] MURPHY R, STRONGIN D R. Surface reactivity of pyrite and related sulfides[J]. Surface Science Reports, 2009, 64(1): 1-45.
[9] MENDIRATTA N K. Kinetic studies of sulfide mineral oxidation and xanthate adsorption[D]. Virginia: Virginia Polytechnic Institute and State University, 2000: 43-49.
[10] AHLBERG E, BROO A E. Oxygen reduction at sulphide minerals. 1. A rotating ring disc electrode (RRDE) study at galena and pyrite[J]. International Journal of Mineral Processing,1996, 46(1/2): 73-89.
[11] AHLBERG E, BROO A E. Oxygen reduction at sulphide minerals. 2. A rotating ring disc electrode (RRDE) study at galena and pyrite in the presence of xanthate[J]. International Journal of Mineral Processing, 1996, 47(1/2): 33-47.
[12] AHLBERG E, BROO A E. Oxygen reduction at sulphide minerals. 3. The effect of surface pre-treatment on the oxygen reduction at pyrite[J]. International Journal of Mineral Processing, 1996, 47(1/2): 49-60.
[13] HAUNG H H, MILLER J D. Kinetics and thermochemistry of amyl xanthate adsorption by pyrite and marcasite[J].International Journal of Mineral Processing, 1978, 5(3):241-266.
[14] PILLAI K C, BOCKRIS J O'M. A quantitative examination of the mixed potential mechanism in mineral flotation[J]. Journal of the Electrochemical Society, 1984, 131(3): 568-579.
[15] BECKER U, HOCHELLA M F Jr. The calculation of STM images, STS spectra, and XPS peak shifts for galena: New tools for understanding mineral surface chemistry[J]. Geochimica et Cosmochimica Acta, 1996, 60(13): 2413-2426.
[16] RAIKAR G N, THURGATE S M. An Auger and EELS study of oxygen adsorption on FeS2[J]. Journal of Physics: Condensed Matter, 1991, 3(12): 1931-1939.
[17] ROSSO K M, BECKER U, HOCHELLA M F. The interaction of pyrite {100} surfaces with O2and H2O: Fundamental oxidation mechanisms[J]. American Mineralogist, 1999, 84(10):1549-1561.
[18] KENDELEWICZ T, DOYLE C S, BOSTICK B C, BROWN G E. Initial oxidation of fractured surfaces of FeS2(100) by molecular oxygen, water vapor, and air[J]. Surface Science, 2004,558(1/3): 80-88.
[19] 王淀佐, 龙翔云, 孙水裕. 硫化矿的氧化与浮选机理的量子化学研究[J]. 中国有色金属学报, 1991, 1(1): 15-23.WANG Dian-zuo, LONG Xiang-yun, SUN Shui-yu. Quantum chemical mechanism on the surface oxidation and flotation of sulfide minerals[J]. The Chinese Journal of Nonferrous Metals,1991, 1(1): 15-23.
[20] SUN W, HU Y H, QIU G Z, QIN W Q. Oxygen adsorption on pyrite (100) surface by density functional theory[J]. Journal of Central South University of Technology, 2004, 11(5): 385-390.
[21] CLARK S J, SEGALL M D, PICKARD C J, HASNIP P J,PROBERT M I J, REFSON K, PAYNE M C. First principles methods using CASTEP[J]. Zeitschrift fur Kristallographie, 2005,220(5/6): 567-570.
[22] SEGALL M D, LINDAN P J D, PROBERT M J, PICKARD C J,HASNIP P J, CLARK S J, PAYNE M C. First-principles simulation: Ideas, illustrations and the CASTEP code[J]. Journal of Physics: Condensed Matter, 2002, 14(11): 2717-2744.
[23] VANDERBILT D. Soft self-consistent pseudopotentials in a generalized eigen value formalism[J]. Physical Review B, 1990,41(11): 7892-7895.
[24] MONKHORST H J, PACK J D. Special points for Brillouin-zone integrations[J]. Physical Review B, 1976, 13(12):5188-5192.
[25] PACK J D, MONKHORST H J. Special points for Brilliouin-zone zntegrations—A reply[J]. Physical Review B,1977, 16(4): 1748-1749.
[26] PRINCE K C, MATTEUCCI M, KUEPPER K. CHIUZBAIAN S G, BARKOWSKI S, NEUMANN M. Core-level spectroscopic study of FeO and FeS2[J]. Physical Review, 2005, 71(8):085102-1-085102-9.
[27] WYCKOFF R W G. Crystal structures [M]. New York:Interscience Publishers, 1963: 85-237.
[28] ROSSO K M, BECKER U, HOCHELLA M F Jr. Atomically resolved electronic structure of pyrite (100) surfaces: An experimental and theoretical investigation with implications for reactivity[J]. American Mineralogist, 1999, 84(10): 1535-1548.
[29] CHATURVERDI S, KATZ R, GUEVREMONT J, SCHOONER M A A, STROGIN D R. XPS and LEED study of a naturally occurring single crystal of pyrite[J]. American Mineralogist,1996, 81(1/2): 261-264.
[30] HUNG A, MUSCAT J, YAROVSDY I, RUSSO S P.Density-functional theory studies of pyrite FeS2(100) and (110)surfaces[J]. Surface Science, 2002, 513(3): 511-524.
[31] 李炳瑞. 结构化学[M]. 北京: 高等教育出版社, 2004: 85-86.LI Bing-rui. Structural chemistry[M]. Beijing: Higher Education Press, 2004: 85-86.
Adsorption of O2on pyrite and galena surfaces
LI Yu-qiong1, CHEN Jian-hua2, LAN Li-hong1,3, GUO Jin4
(1. School of Chemistry and Chemical Engineering, Guangxi University, Nanning 530004, China;2. College of Resources and Metallurgy, Guangxi University, Nanning 530004, China;3. College of Chemistry and Ecoengineering, Guangxi University for Nationalities, Nanning 530006, China;4. College of Physics Science and Engineering, Guangxi University, Nanning 530004, China)
The adsorption of oxygen molecule (O2) on the pyrite and galena surfaces was studied using density functional theory (DFT). The calculated results show that the surface relaxation of pyrite and galena is small. O2dissociates after adsorption on the pyrite and galena surfaces, and the adsorption energy of O2on pyrite is much lower than that on the galena. On the pyrite surface, oxygen atom (O) bonds with sulfur (S) and iron (Fe) atoms and the electrons are transferred from Fe and S atoms to O, and the reactions are mainly S 3p, O 2p and Fe 3d states involved, forming the d→p back bonding between Fe and O. While on the galena surface, oxygen atom only bonds with sulfur atom, and the reactions are mainly S 3p, O 2p and Pb 6p states involved, without forming d→p back bonding. The bonded Fe and O atoms are spin-polarized after the adsorption of O2, while the galena surface atoms and adsorbed O atom are still low-spin states.
pyrite; galena; O2adsorption; density functional theory
TD923
A
1004-0609(2012)04-1184-11
国家自然科学基金资助项目(50864001)
2011-01-20;
2011-07-11
陈建华,教授,博士;电话:0771-3232200;E-mail: jhchen1971@sina.com
(编辑 龙怀中)
