选择性降低卷烟烟气中CO和NOx的钙钛矿型催化剂研究
2012-11-22谢国勇银董红刘建福钱晓春
谢国勇,银董红,刘建福,钱晓春
(湖南中烟工业有限责任公司技术中心,中国 长沙 410007)
一氧化碳(CO)和氮氧化物(NOx)是卷烟烟气中的主要有害物质[1-2], 严重危害人体健康[3-5].催化氧化法以金属或金属氧化物为催化剂,将烟气中CO选择性氧化为CO2,并将NOx选择性还原为N2,被认为是最有效和最有应用前景的选择性降害途径之一.
目前,研究较多的是在卷烟滤棒区域实现CO的低温选择性催化氧化.国内外相继开发了担载在活性炭和膨润土混合物上的Pd-MnO2、Pd-Cu、Pd-Cu-V催化材料[6-8];以碱金属为助剂的Pt-FeO3/TiO2、Pt-V2O5-Fe2O3/Al2O3催化剂[9];Pt-V2O5-Fe2O3/Al2O3催化剂[10],长沙卷烟厂应用纳米贵金属复合滤棒,成功研制出低焦油、低CO白沙“和”牌卷烟产品[11-12].但是,昂贵的价格在很大程度上限制了贵金属烟用催化剂的广泛应用.因此,低成本、高活性烟用催化剂的研发显得尤为重要.
根据卷烟的燃烧特性,在燃吸过程中,卷烟的热解区和燃烧区为200~900 ℃的中高温区域[2,13].此温度条件有利于过渡金属和稀土金属氧化物等低成本中高温催化剂对一氧化碳等的催化脱除.因此,将中高温催化剂添加于烟草薄片、烟丝及卷烟纸可望实现烟气中CO和NOx等有害物质的有效脱除.本文针对卷烟的吸燃特性及烟气的复杂成分,设计了低成本且具有高氧化还原催化活性和强水热稳定性的纳米级La0.7Ln0.3Fe1-yMyO3钙钛矿型复合氧化物催化剂(其中Ln为稀土金属离子,M为过渡金属),在催化氧化CO的同时,将CO催化氧化和NOx催化还原反应耦合,于卷烟燃烧区和热解区实现卷烟烟气中CO和NOx等有害物质的同时高效脱除.
1 实验部分
1.1 催化剂制备
采用柠檬酸为配体的溶胶-凝胶法制备钙钛矿型复合氧化物.将按化学计量比混合的硝酸盐混合物溶于超纯水中,然后加入稍大于金属离子总量的柠檬酸水溶液并充分搅拌.混合溶液于70 ℃温度条件下搅拌蒸发水份,直至形成透明溶胶(Sol),再放置5~6 h形成流动性较差的凝胶(Gel),接着将所得凝胶于120 ℃温度条件下干燥8 h得到疏松多孔干凝胶,将此干凝胶研磨后于不同温度下焙烧,即得所需样品.
在催化剂制备过程中,以LaFeO3钙钛矿复合氧化物为模板,采用双位取代的方案,亦即A位的稀土元素La被Ln部分取代的同时,B位元素Fe被某过渡元素M部分取代,优化催化剂组成.所得催化剂表示为La1-xLnxFe1-yMyO3(x,y为取代值).
1.2 催化剂活性评价
催化剂活性评价在一固定床石英玻璃微型反应器内进行(内径6 mm、长度650 mm).常压操作,催化剂用量为50 mg,模拟烟气流量为100~1 000 mL/min,烟气组成为:4.0%CO+0.1%NO+(0~10.0)%O2+(0~10.0)%CO2+(0~3.0)%H2O,He为载气.原料气和反应尾气中CO、NO、NO2、O2、CO2等的分析采用气相色谱(Agilent 6890N,碳分子筛色谱填充柱,TCD检测器)和燃气分析仪(KM96006, Quintox, Kane International Ltd)在线检测.催化剂催化活性对比用CO转化率为10%、50%、90%所对应的温度T10%、T50%和T90%表示.其中T10%表示为催化起燃温度.
1.3 催化剂表征分析
TG/DSC分析:采用Netsch STA449c型热分析仪对120 ℃烘干的凝胶样品进行热分析,在空气气氛下以10 ℃/min的速度升温,升温范围为25~900 ℃.采用日本理学D/max 2550型X射线仪(Cu Kα辐射,Ni 滤波,管电压40 kV,管电流300 mA)对样品进行物相分析.BET比表面分析:通过Micromeritics ASAP-2010 型自动吸附仪测定样品对氮的吸附等温线(73 K) ,用BET 法计算样品的比表面积.粒子大小测定及形貌观察在JEOL JEM-200S型透射电镜上进行.
1.4 烟草薄片、试制烟的制备及其检测
采用造纸法制备烟草薄片.将一定量催化剂研磨至200目后与松木桨液充分混合,静置8~10 h以使催化剂充分粘附在纤维上,然后将此混合物真空抽滤成薄片,并置于120 ℃烘箱内烘烤2 h,即得烟草薄片.将制得的烟草薄片切成1.1×15 mm的小块,并与烟丝充分混合,于Huani Protos 70型卷烟机上卷制试制烟.然后在Filtrona SM400型直线式吸烟机上对试制烟进行抽吸,每组试样占用5个通道,每个通道抽吸4支烟,烟气中CO含量按YC/T30-1996 《卷烟烟气气相中一氧化碳的测定——非散射红外法》测定, NOx采用离子色谱(Dionex 2500)检测.
2 结果与讨论
2.1 催化剂制备条件研究
在以柠檬酸络合溶胶凝胶法制备钙钛矿复合氧化物过程中,对样品平均粒径及性能影响较大的制备条件主要包括:总金属离子浓度、柠檬酸用量、形成溶胶凝胶温度、干燥条件及焙烧温度等.其中,焙烧条件(焙烧温度和焙烧时间)对样品的影响非常重要.因此,在本部分重点考察焙烧条件对样品物化性能的影响.
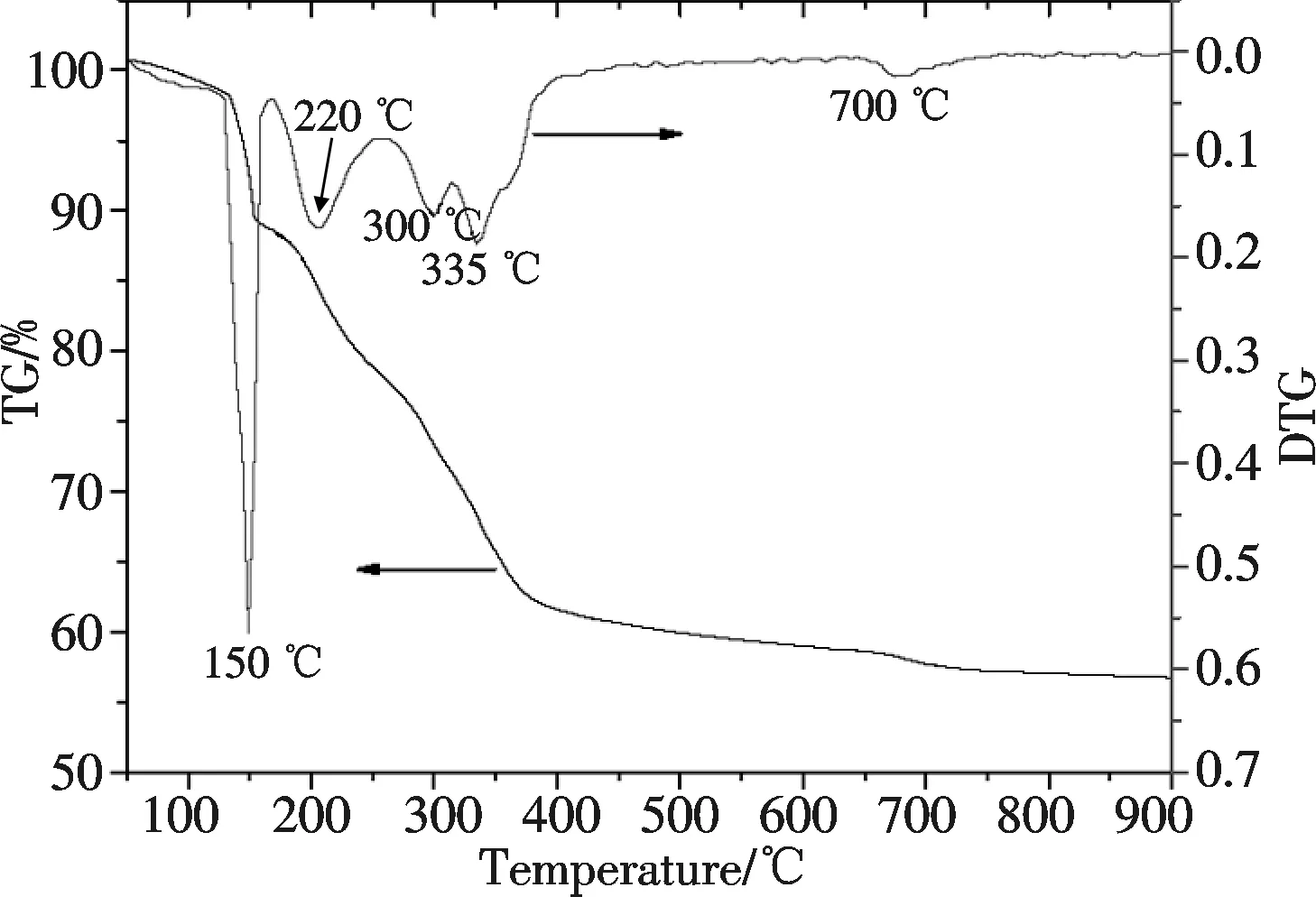
图1 La0.7Ln0.3FeO3干凝胶的TG-DTG分析
2.1.1 热分析结果
首先采用热分析方法研究所制备的La0.7Ln0.3FeO3干凝胶的热分解过程,以确定最佳的焙烧温度.如图1所示,溶胶-凝胶法制备的干凝胶在150 ℃左右有一明显的失重和放热峰,这是因为柠檬酸发生燃烧,这一过程进行迅速并放出大量热,在此过程中NO3-也部分分解.随着温度升高,样品在150 ℃~400 ℃温度范围内发生缓慢失重,并分别于220 ℃、300 ℃和335 ℃出现失重峰,在此温度范围内,剩余的NO3-被逐步分解并生成钙钛矿前驱体.温度进一步升高至700 ℃左右,样品又出现一较小的失重峰.这一微小失重峰是成矿过程中氧空穴等的形成所致[14].因此,从热分析数据可推断La0.7Ln0.3FeO3钙钛矿的晶化温度为700 ℃左右.
2.1.2 XRD物相和BET比表面分析
根据上述热分析结果,将120 ℃干凝胶在静态空气下分别于500 ℃、700 ℃、800 ℃、900 ℃焙烧2 h,然后进行XRD和比表面分析.图2为120 ℃干凝胶分别在不同温度下于空气中焙烧2 h所得试样的XRD表征结果.由该图可见,500 ℃焙烧样品呈现较弱的XRD衍射峰,基本为无定形结构.焙烧温度升至700 ℃,样品出现较强的LaFeO3钙钛矿XRD衍射峰(与JCPDS卡片37-1493相吻合),且随着焙烧温度进一步升高,衍射峰强度增大,但当焙烧温度达到900 ℃以后,样品出现Fe2O3的衍射峰(与JCPDS卡片25-1402相吻合),这说明高温焙烧可能导致LaFeO3钙钛矿结构的破坏.从图2还可以看出,700 ℃以上焙烧样品中均存在LnOx的XRD衍射峰,这说明在成矿过程中有部分LNOx晶相析出.图3显示500 ℃ 2 h焙烧条件下,La1-xLnxFeO3样品XRD衍射峰随Ln掺杂因子x的变化关系.由图可见,当x为0和0.1时,样品呈现单一的LaFeO3钙钛矿XRD衍射峰,随着x增至0.2、0.3、0.4样品中检测到LnOx衍射峰,且其强度随着x的增大而增强.图3的结果说明Ln离子在LaFeO3中的容限因子小于0.2,与文献相符[15].而后面的催化活性评价结果显示,掺杂因子x为0.3的La0.7Ln0.3FeO3具有最高的催化活性,这可能是因为LnOx晶相中的Lnn+与钙钛矿中其他金属离子具有良好的协同作用[16].

(a)500 ℃ 2 h,(b)700 ℃ 2 h,(c)800 ℃ 2 h,(d)900 ℃ 2 h (a)x=0,(b)x=0.1,(c)x=0.2,(d)x=0.3,(e)x=0.4图2 La0.7Ln0.3FeO3的XRD衍射峰随焙烧温度的变化关系 图3 La1-xLnxFeO3的XRD衍射峰随掺杂因子x的变化关系
对不同温度焙烧样品的BET比表面测试结果(表1)分析表明,随着焙烧温度升高,样品比表面逐渐降低,这说明晶化的粒子随着焙烧温度的升高而逐渐聚集长大,900 ℃焙烧样品的比表面积降低至6.51 m2/g.表1同时还给出了样品于700 ℃焙烧6 h的比表面积,在相同温度条件下,焙烧温度自2 h增至6 h,其比表面积稍有降低.以上结果表明样品在700 ℃焙烧2 h可以得到稳定的LaFeO3钙钛矿结构,且具有较大的比表面积.

表1 不同焙烧条件LaFeO3钙钛矿的BET比表面积表征结果
2.1.3 SEM和TEM分析
图4a,b分别为700 ℃焙烧2 h所得La0.7Ln0.3FeO3钙钛矿的SEM和TEM照片.由图4a可见,该催化材料表面含有丰富的类似蜂窝状的孔道结构,这是由于该溶胶凝胶制备方法中添加了有机络合剂柠檬酸.样品孔道形成可能缘于以下两个方面:一方面是在制备过程中发泡形成的蓬松多孔结构;另一方面是在焙烧阶段柠檬酸燃烧而留下较多的孔道结构.从图4b所示的TEM照片可估算所得催化剂颗粒的平均粒径约为50 nm,该结果表明,采用柠檬酸络合溶胶凝胶法可制备纳米级钙钛矿材料.

图4 700 ℃焙烧2 h所得La0.7Ln0.3FeO3钙钛矿的SEM(a)和TEM(b)照片
2.1.4 焙烧温度对催化剂活性的影响
对于钙钛矿复合金属氧化物,较高的晶化程度和比表面是获得高催化活性的前提条件.图5为不同温度焙烧所得La0.7Ln0.3FeO3钙钛矿样品的CO催化氧化活性评价结果.从图中可以看到,500 ℃焙烧样品的T50%为330 ℃,随着焙烧温度升高至700 ℃,样品的T50%降至295 ℃,而当焙烧温度进一步升高至800 ℃,样品的T50%又升至310 ℃,亦即700 ℃焙烧样品具有最高的CO催化氧化活性.结合前述的XRD和比表面分析结果,随着焙烧温度的升高,La0.7Ln0.3FeO3钙钛矿的晶化程度逐渐提高,而其比表面积逐渐降低,在700 ℃焙烧2 h条件下,样品可形成稳定的LaFeO3钙钛矿结构,且具有较大的比表面积,因此具有较高的催化活性.
2.2 A、B位部分取代对LaFeO3钙钛矿催化活性的影响
图6a~d为LaFeO3钙钛矿及其La3+(A位)被Lnn+部分取代后La1-xLnxFeO3(x=0.1-0.4)的CO催化氧化活性随反应温度的变化关系.LaFeO3(图6a)的起燃温度为310 ℃ (T10%),当反应温度升至375 ℃时CO转化率达到50% (T50%),继续升温至440 ℃,CO转化率达到90%(T90%).当La3+被Lnn+部分取代后,催化剂的催化活性随着取代值x自0增至0.3而逐渐升高,La0.7Ln0.3FeO3(图6c)的T10%、T50%和T90%分别降至228 ℃、300 ℃和360 ℃.但当取代值x进一步增加至0.4时,催化剂(图6d)的催化氧化活性反而下降.

反应条件:CO 4%,O2 4%,样品 50 mg,GHSV 4.8×105 L·kg-1·h-1图5 焙烧温度对La0.7Ln0.3FeO3钙钛矿催化活性的影响

(a) LaFeO3, (b) La0.8Ln0.2FeO3, (c) La0.7Ln0.3FeO3, (d) La0.6Ln0.4FeO3, (e) La0.7Ln0.3Fe1-yMyO3反应条件:CO 4%, O2 4%, 样品50 mg, GHSV 4.8×105 L·kg-1·h-1图6 A、B位替代对LaFeO3钙钛矿复合金属氧化物CO+O2催化氧化活性的影响
对于ABO3型钙钛矿复合金属氧化物,其催化活性主要由B位离子决定,A位离子主要通过控制活性组分B的原子价态和分散状态而起稳定结构的作用.A位离子本质上不直接参与反应,但是若被价态不同的其他离子取代,就会引起B位离子价态的变化,使得不寻常价态离子增多,同时也可能造成晶格缺陷,从而改变晶格氧的化学位[17-18],这些是Lnn+掺杂导致催化活性增强的主要原因.由前述图2、图3的XRD表征可知,Lnn+在La1-xLnxFeO3内的容限因子x为0.2,当x超过0.2时,有LnOx晶相析出,图6c La0.7Ln0.3FeO3高催化活性可能源于LnOx晶相中的Lnn+与钙钛矿中其他金属离子具有良好的协同作用,但过量Lnn+掺杂可能会导致钙钛矿结构的破坏,从而影响催化活性.
当La0.7Ln0.3FeO3钙钛矿的Fe3+(B位)被某过渡金属元素MⅡ部分取代后,如图7e所示,催化剂的活性温度T10%、T50%和T90%分别自La0.7Ln0.3FeO3的228 ℃、300 ℃和360 ℃降至La0.7Ln0.3Fe1-yMyO3的175 ℃、255 ℃和320 ℃,催化活性大幅提高.当La0.7Ln0.3FeO3钙钛矿的Fe3+离子被低价态M2+部分取代后,直接导致不稳定的高价态Fe4+增多,从而增强其氧化还原性能.而Fe4+可能充当了催化剂活性位的角色.
由图6分析可知,当LaFeO3钙钛矿的La3+(A位)和Fe3+(B位)同时被稀土金属Ln和过渡金属MⅡ部分取代后,其催化活性大幅提高,其中La0.7Ln0.3Fe1-yMyO3表现出最高的CO催化氧化活性,被选为最佳催化剂组成.
2.3 La0.7Ln0.3Fe1-yMyO3钙钛矿催化剂同时脱除CO和NO
图7为La0.7Ln0.3Fe0.9M0.1O3钙钛矿复合金属氧化物催化剂在100~600 ℃温度范围内同时脱除模拟烟气中CO和NO的反应催化活性评价结果,实验分别在有氧((1) CO+NO+O2)和无氧((2) CO+NO)两种气氛中进行.从图7a可以看出,在(1) CO+NO+O2反应气氛下,CO催化氧化的T10%、T50%和T90%分别为124 ℃、153 ℃和181 ℃,而NO基本无转化.在(2) CO+NO反应气氛下,CO催化氧化和NO催化还原的T10%、T50%和T90%分别为225 ℃、335 ℃和438 ℃.图7b为在CO+NO反应气氛下,反应组分CO、NO以及反应产物N2和CO2随反应温度的变化关系.实验过程中,未检测到N2O的产生.
在钙钛矿复合金属氧化物上,模拟烟气中CO、NO、O2等组分之间存在CO的催化氧化、CO还原NO以及NO的催化分解等反应.在这些反应中氧空位起着非常重要的作用,由于氧化剂O2和NO相互争夺氧空位而导致上述3个反应之间存在竞争.在(1) CO+NO+O2反应气氛下,催化剂表面氧空位首先从气相中吸附氧,吸附氧与阳离子的结合比较弱,形成了活性氧物种,并与气相中的CO发生反应.因此,CO催化氧化反应占绝对优势,而CO还原NO反应受到抑制,如图7(a)所示.在(2) CO+NO反应气氛下,CO首先夺取氧化物上的晶格氧,形成氧空穴,然后氧空缺吸附NO,由于这种吸附属化学吸附,吸附的NO分子中的氧原子与邻近的金属原子形成了化学键,同时减弱了氧原子和氮原子之间的共价键,这种被削弱的共价键很容易断裂,结果形成了新的晶格氧,同时产生N2分子逸出.因此,在缺氧条件下,主要发生CO还原NO反应.

反应条件:CO 4%, NO 4%, O2 4%(若使用), 样品50 mg, GHSV 4.8×105 L·kg-1·h-1图7 La0.7Ln0.3Fe1-yMyO3钙钛矿CO+O2和CO+NO催化活性评价结果
根据上述对La0.7Ln0.3Fe1-yMyO3钙钛矿催化剂同时脱除模拟烟气中CO和NO实验结果的分析,如将所制备的La0.7Ln0.3Fe1-yMyO3钙钛矿复合氧化物添加于卷烟薄片,在卷烟吸燃过程中,可以于相对富氧的热解区实现CO的催化氧化脱除.在此区域,钙钛矿复合氧化物担当催化剂的角色;而在缺氧的燃烧区,通过CO还原NO反应耦合CO催化氧化和NO催化还原,实现NO的有效脱除.同时,钙钛矿复合氧化物以及稀土金属氧化物的贮氧功能还可促进CO的氧化,这可以在一定程度上补偿燃烧区的缺氧状况.在此区域,钙钛矿复合氧化物担当催化剂和氧化剂的双重角色.因此,钙钛矿复合氧化物经过卷烟热解区和燃烧区的催化和氧化作用,有望实现卷烟烟气中CO和NO等有害物质的有效脱除.
2.4 卷烟烟气测试结果
对添加La0.7Ln0.3Fe1-yMyO3钙钛矿催化剂的试制烟进行烟气检测,并与不含催化剂空白对比样的烟气检测结果进行对比,结果如表2所示.分别考察了钙钛矿质量分数为0.8%和2.0%试制烟对烟气中CO和NOx的脱除效果.往卷烟中添加0.8%的La0.7Ln0.3Fe1-yMyO3钙钛矿催化剂,烟气中的CO、NOx和总粒相物分别自14.29 mg/支、36.42 μg/支、16.12 mg/支降至12.84 mg/支、32.31 μg/支、15.32 mg/支,降幅为10.1%、11.3%、4.9%,当La0.7Ln0.3Fe1-yMyO3钙钛矿催化剂添加量增至2.0%,CO、NOx和总粒相物进一步降低至12.02 mg/支、31.34 μg/支、14.94 mg/支,降幅达到16.0%、15.9%、7.3%,其中总粒相物的降幅远低于CO和NOx的降幅.同时,烟支的抽吸口数略有降低,这可能是因为催化剂对卷烟有一定的助燃作用,而总粒相物的降低也可能与此有关.以上实验结果说明,添加2.0%La0.7Ln0.3Fe1-yMyO3钙钛矿催化剂于卷烟中,可选择性地降低烟气中CO和NOx的含量,并且对卷烟的吸阻和抽吸口数影响不大.
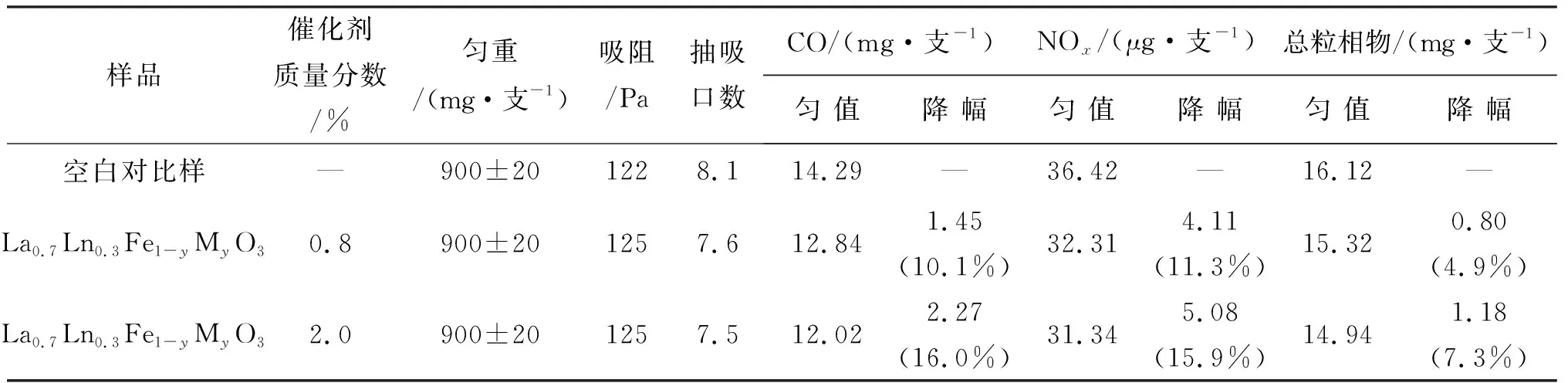
表2 卷烟测试结果
3 结论
(1) 柠檬酸络合溶胶-凝胶法是一种合适的纳米级钙钛矿复合氧化物制备方法,可以在较低焙烧温度条件下形成钙钛矿结构晶型.
(2) LaFeO3是一种性能优良的CO催化氧化催化剂,当La3+(A位)和Fe3+(B位)分别被稀土金属Ln和过渡金属M部分取代后,其催化活性大幅提高.其中,La0.7Ln0.3Fe1-yMyO3具有最高的CO催化氧化和NO催化还原活性.
(3) 将2%质量分数的La0.7Ln0.3Fe1-yMyO3钙钛矿复合氧化物添加于卷烟中,可有效降低卷烟烟气中CO和NOx的质量,并且相对于焦油、烟碱等粒相物的降低具有很高的选择性.
参考文献:
[1] DAVIS D L, NIELSEN M T.烟草——生产,化学和技术[M].国家烟草专卖局科技教育司和中国烟草科技信息中心组织编译.北京:化学工业出版社,2003:380-410.
[2] BAKER R R. Product formation mechanisms inside a burning cigarette [J]. Prog Energy Combust Sci, 1981,7(2):135-153.
[3] 周君富,郭芳珍,钱志君.一氧化氮等自由基对吸烟者损害效应的研究[J].中国公共卫生, 1983,13(2):90-921.
[4] 陈清江,翟海滨,周君富.吸烟者烟雾中一氧化氮等自由基对机体抗氧化能力的影响[J].中华航海医学与高气压医学杂志, 2001,8(3):151-1541.
[5] 赵保路. 再论香烟烟气中一氧化氮对人体的作用[J]. 北京烟草, 2000(3):20-21.
[6] 鹤田邦弘,牧正雄.一种卷烟净滤器:日本,59/029036[P]. 1981-02-14.
[7] 市濑茂男,斌森健一郎.卷烟中一氧化碳去除剂:日本,60/224483[P]. 1985-11-08.
[8] 松下肇.一氧化碳去除触酶制造方法:日本,56/015683[P]. 1984-02-16.
[9] TOOLEY P A, KOLTS J H. Catalyst for oxidation of carbon monoxide: US, 4940686[P]. 1990-07-10.
[10] ELLIOTT D J, KOLTS J H. Catalyst composition for the oxidation of carbon monoxide: US, 4956330[P]. 1990-09-18.
[11] 吕功煊,刘建福,越明月,等.降低卷烟主流烟气中一氧化碳释放量的催化剂及其制备方法和应用:中国,03121750.8[P]. 2008-06-25.
[12] 吕功煊,聂 聪,赵明月,等.应用含纳米贵金属催化材料降低卷烟烟气中CO技术研究[J].中国烟草学报, 2003,9(3):18-27.
[13] BAKER R R. A review of pyrolysis studies to unravel reaction steps in burning tobacco [J]. J Anal Appl Pyrolysis, 1987,11(3-4):555-573.
[14] TEJUCA L G, FIERRO J L G. Properties and application of perovskite-type oxides [M]. New York:Marcel Dekker, Inc, 1992.
[15] 崔梅生,李明来,张顺利,等.钙钛矿催化材料La1-xLnxCoO3的制备、表征及甲烷燃烧催化性质[J].中国有色金属学报, 2004,14 (9):1580-1584.
[16] LI H Y,GE S Q. Application of nano-material of rare earth for the purification of automobile exhaust gases [J]. New Chem Mater, 2002,30(8):29-31.
[17] TANAKA H, MISONO M. Advances in designing perovskite catalysts[J]. Curr Opin Solid State Mater Sci, 2001,5(5):381-387.
[18] LIBBY W F. Promising catalyst for auto exhaust [J]. Science, 1971,171(3970):199-500.
