新型3-甲基黄嘌呤衍生物的合成
2012-11-21张月成赵继全
白 洁, 张月成, 赵继全
(河北工业大学 化工学院,天津 300130)
3-甲基黄嘌呤(6a)是合成嘌呤类衍生物的重要医药中间体,如MRS 1754, DMPTX、舒安灵等[1~4]。研究发现黄嘌呤类化合物的8-C被苯基取代能大大提高腺苷受体的亲和力,在治疗心脑血管,平喘,利尿等方面起着重要作用[5]。
目前6a的合成路线主要有两条[6]。路线一:以甲基脲与氰乙酸为起始原料,在醋酸酐催化下生成3-甲氰乙酰脲(1);1经环合和亚硝化制得3-甲基-4-氨基-5-亚硝基脲嘧啶(3); 3经还原、酰化、闭环及酸化合成6a。路线二:以1-甲基-4-氯咪唑为起始原料,经亚化、取代、水解、还原、重排及缩合合成6a。路线一具有的原料易得、价格便宜、反应条件温和及操作简单等优点,更适合工业化生产。
本文采用路线一合成了6a及其衍生物(6b~6f, Scheme 1),其中6c~6f为新化合物,其结构经1H NMR和FT-IR表征。并对合成工艺进行了优化,提高了收率。
1 实验部分
1.1 仪器与试剂
XT-4型双目显微熔点仪(温度未校正);Bruker AC-P 400型核磁共振仪(DMSO-d6为溶剂,TMS为内标);Bruker Vector 22型傅立叶变换红外光谱仪(KBr压片);HITACHI L-2130型高效液相色谱仪。
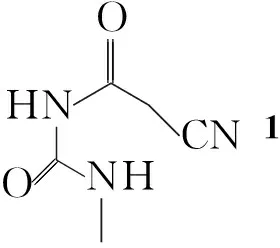

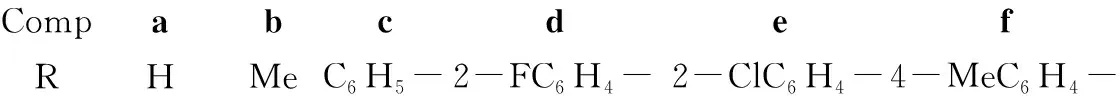
CompabcdefRHMeC6H5-2-FC6H4-2-ClC6H4-4-MeC6H4-
Scheme1
有机酸(Ⅰa~Ⅰf),分析纯,国药集团化学试剂有限公司;实验用水,去离子水;其余所用试剂均为分析纯。
1.2 合成
(1) 3-甲基-4-氨基脲嘧啶(2)的合成[7,8]
在反应瓶中加入氰乙酸17.01 g(200 mmol)的醋酸酐(33 mL)溶液,搅拌下于室温分批加入甲基脲29.63 g(400 mmol),于90 ℃反应1 h。于60 ℃减压蒸除乙酸;降至室温,过滤,滤饼用水洗涤多次,真空干燥得白色粉末1 27.18 g,收率96.4%, m.p.204 ℃~205 ℃(74%, 204 ℃~205 ℃[7])。
在反应瓶中加入混合溶液[V(10%KOH) ∶V(22%NaOH)=1 ∶2]16 mL,搅拌下于45 ℃~55 ℃分批加入114.10 g(100 mmol),加毕,于50 ℃反应2.5 h。降至45 ℃加去离子水23 mL,降至室温,用2 mol·L-1盐酸调至pH 6。静置过夜,过滤,滤饼真空干燥得白色固体2 12.61 g,收率89.5%(88%[8]),纯度99.7%(HPLC,下同);1H NMRδ: 10.39(s, 1H, NH), 6.56(s, 2H, NH2), 4.50(s, 1H, CH), 3.32(s, 3H, CH3); IRν: 3 365, 3 181, 1 625, 1 510, 1 456, 1 387, 1 300, 1 234, 1 187, 1 064, 987, 854, 775, 732, 657, 537 cm-1。
(2)3的合成[9]
在反应瓶中加入浓硫酸1.8 mL和水30 mL,搅拌下于50 ℃滴加混合溶液[2 11.28 g(80 mmol)+亚硝酸钠5.52 g(80 mmol)+水(50 mL)],滴毕,用10%硫酸调至pH 1.5~2.0,反应至终点(KI试纸检测变蓝)。降至室温,过滤,滤饼用水洗至中性,真空干燥得粉红色固体312.93 g,收率95.1%(85%[9]),纯度99.8%;1H NMRδ: 10.49(s, 1H, NH), 6.15(s, 2H, NH2), 3.33(s, 3H, CH3); IRν: 3 446, 3 273, 3 226, 1 706, 1 657, 1 569, 1 515, 1 393, 1 273, 1 110, 873, 778, 703, 574 cm-1。
(3) 3-甲基-4,5-二氨基脲嘧啶(4)的合成[10]
在高压釜中加入3 8.5 g(50 mmol),骨架Ni 1.4 g及去离子水70 mL,开搅拌充氮气置换釜内空气;充氢气置换,待充氢气至0.5 MPa时,于50 ℃反应至终点(氢气消耗量接近理论氢气消耗值且氢气压力表数值不变为止)。降至室温,过滤,滤饼真空干燥得黄色固体47.02 g,收率92.0%(90%[10]),纯度99.2%;1H NMRδ: 10.51(s, 1H, NH), 6.10(s, 2H, NH2), 2.81(s, 2H, NH2), 3.33(s, 3H, CH3); IRν: 3 355, 3 314, 3 262, 3 154, 1 625, 1 589, 1 509, 1 388, 1 266, 1 130, 965, 896, 830, 751, 682, 564 cm-1。
(4)6a~6f的合成[7,11]
在反应瓶中加入4 7.00 g(40 mmol)和去离子水70 mL,搅拌使其溶解;于50 ℃加甲酸(Ⅰa)4.6 mL,滴毕,回流反应1 h。用30%NaOH溶液调至pH 11~12,回流反应30 min。蒸干溶剂得橙红色固体,用异丙醇重结晶得得淡黄色固体。用水10 mL溶解,于45 ℃用2 mol·L-1盐酸调至中性,冷却至室温,过滤,滤饼真空干燥得6a。用类似的方法合成6b。
在反应瓶中加入4 1.09 g(6.79 mmol)和苯甲酸(Ⅰc)1.71 g(14.01 mmol),搅拌下于140 ℃恒温反应4 h。冷却室温,加甲醇8 mL,过滤,滤饼干燥得黄色固体5c。用类似的方法合成黄色固体5d~5f。
将5c用混合溶剂[V(2 mol·L-1NaOH) ∶V(甲醇)=7 ∶1]10 mL溶解,搅拌下回流反应6 h。冷却至室温,用2 mol·L-1盐酸调至中性,过滤,滤饼依次用甲醇、水洗涤,真空干燥得6c。用类似的方法合成6d~6f。
6a: 淡黄色固体,收率85.5%(62%[7]),纯度99.6%;1H NMRδ: 13.48(s, 1H, NH), 10.08(s, 1H, NH), 8.01(s, 1H, CH), 3.37(s, 3H, CH3); IRν: 3 135, 3 085, 2 985, 2 825, 1 700, 1 564, 1 441, 1 414, 1 282, 1 251, 1 165, 976, 934, 847, 798, 745, 615, 532 cm-1。
6b: 白色固体,收率84.2%(62%[7]),纯度99.6%;1H NMRδ: 13.50(s, 1H, NH), 10.18(s, 1H, NH), 3.33(s, 3H, CH3), 2.54(s, 3H, CH3); IRν: 3 142, 3 025, 2 927, 1 722, 1 565, 1 432, 1 454, 1 267, 1 241, 1 167, 986, 932, 852, 796, 732, 711, 630, 502 cm-1。
6c: 淡黄色固体,收率70.0%,纯度99.7%;1H NMRδ: 13.70(s, 1H, NH), 11.12(s, 1H, NH), 8.11~8.13(m, 2H, ArH), 7.51~7.48(m, 3H, ArH), 3.43(s, 3H, CH3); IRν: 3 164, 3 037, 2 824, 1 687, 1 524, 1 470, 1 418, 1 276, 1 201, 976, 845, 709, 619, 544 cm-1。
6d: 黄色固体,收率73.5%,纯度99.9%;1H NMRδ: 13.73(s, 1H, NH), 11.15(s, 1H, NH), 7.73~7.46(m, 4H, ArH), 3.41(s, 3H, CH3); IRν: 3 553, 3 454, 3 173, 3 102, 3 039, 2 835, 1 705, 1 672, 1 621, 1 554, 1 519, 1 476, 1 450, 1 283, 1 246, 1 173, 1 090, 825, 768, 738, 695, 550 cm-1。
6e: 黄色固体,收率72.2%,纯度99.7%;1H NMRδ: 13.70(s,1H, NH), 11.07(s, 1H, NH), 7.98~7.33(m, 4H, ArH), 3.42(s, 3H, CH3); IRν: 3 480, 3 367, 3 239, 3 063, 2 990, 2 860, 2 814, 1 705, 1 629, 1 584, 1 508, 1 417, 1 310, 1 177, 1 044, 1 026, 887, 746, 709, 564 cm-1。
6f: 黄色固体,收率65.1%,纯度99.8%;1H NMRδ: 13.69(s, 1H, NH), 11.09(s, 1H, NH), 8.02~8.00(m, 2H, ArH), 7.32~7.30(m, 2H, ArH), 3.43(s, 3H, NCH3), 2.36(s, 3H, ArCCH3); IRν: 3 437, 3 172, 3 030, 2 954, 2 823, 1 685, 1 531, 1 478, 1 419, 1 363, 1 271, 1 111, 1 025, 831, 728, 546 cm-1。
2 结果与讨论
2.1 合成工艺优化
(1) 2的合成
对2合成中的缩合、闭环工艺优化中发现,缩合反应温度对2收率影响最大,结果见表1。由表1可以看出,随着反应温度的升高,2的收率明显提高,反应温度为90 ℃时收率最高(96.4%);再提高温度,收率影响不大;温度超过100 ℃时氰乙酸开始分解,致使收率下降。最佳缩合反应温度为90 ℃。

表1 缩合反应温度对2收率的影响*Table 1 Effect of condensation temperature on yield of 2
*反应时间1 h,其余反应条件同1.2(1)
(2) 3的合成
在3的合成中,对文献[11]方法的亚硝化方式进行了改进,采用将亚硝酸钠与2的水溶液滴加到硫酸溶液中。亚硝酸钠与硫酸反应生成亚硝酸后能及时与2发生亲电加成,亚硝酸钠不会以氮氧化物的形式溢出,反应更充分,收率95.1%,高于文献收率(85%)。
(3) 4的合成
在4的合成中发现,采用催化加氢的方法比用连二亚硫酸钠[10]方法的还原效果更好,不仅反应时间由28 h缩短至4 h,收率从85%提高至90%,纯度由93.6%提高至99.2%,而且后处理简单,Ni可以回收处理后循环使用,降低成本。
(4) 6的合成

表2 Ⅰ的取代基位置和反应时间对6收率的影响Table 2 Effect of substituting position of Ⅰ and reaction time on yield of 6
在6c~6f的合成中发现苯甲酸的邻位为吸电子基团(Ⅰd和Ⅰe)时有利于反应进行,而对位为给电子基团(Ⅰf)时则不利于反应进行。Ⅰd和Ⅰe与4反应合成6d和6e所需反应时间缩短,且收率提高2%;而Ⅰf所需时间变长且收率降低约5%(表2)。
[1] Maria Carmen Balo, Jose Brea. Synthesis and pharmacological evaluation of novel 1- and 8-substituted-3-fururyl xanthines as adenosine receptor antagonists[J].Med Chem,2009,17:6755-6760.
[2] Ma Dong. Synthesis of 8-substituted xanthines via 5,6-diaminouracils:An efficient route to A2Aadenosine receptor antagonists[J].Tetrahedron Letters,2008,49:4633-4635.
[3] Maria Isabel Nieto. Synthesis of novel 1-alkyl-8-substituted-3-(3-methoxypropyl) xanthines as putative A2Areceptor antagonists[J].Med Chem,2009,17:3426-3432.
[4] 张词侠,张桂林,李志勇. 已酮可可碱的药理特性和治疗用途的研究进展[J].中国临床药理学杂志,2008,24(1):74-77.
[5] Jacobson K A. 1,3-Dialkylxanthine derivatives having high potency as antagonists at human A2Badenosine receptors[J].Drug Dev Res,1999,47:45-53.
[6] A V Gulevskaya. Synthesis ofN-substituted xanthines(Review)[J].Chemistry of Heterocyclic compounds,1991,27(1):1-23.
[7] Per G Kjellin. 3,8-Dialkylxanthines[P].US 4 546 182,1985.
[8] Russell D Drinkard. Preparation of 4-aminouracil[P].US 2 804 459,1957.
[9] 刘胜高. 3-甲基-4-亚氨基紫脲酸的合成[J].山东化工,2008,37(1):6-8.
[10] Barbara Ann Spicer. Synthesis of 8-substituted xanthines[P].US 6 180 791,2001.
[11] David Beer. A solid-phase approach towards the synthesis of PDE5 inhibitors[J].Med Chem Lett,2002,12:1973-1976.
