山羊PHGPx基因干扰质粒构建及其抑制效应评价
2012-10-26张春香施力光荀文娟王茜任有蛇岳文斌
张春香,施力光,荀文娟,王茜,任有蛇,岳文斌
(1.山西农业大学 动物科技学院,山西 太谷030801;2.中国热带农业学院 热带牧草和畜禽研究所,海南 儋州571737)
硒蛋白PHGPx作为谷胱甘肽过氧化物酶家族的中重要成员,不仅能特异清除磷脂氢过氧化氢物,保护生物膜系统免受氧化应激损伤,近年还发现PHGPx可通过信号转传导刺激细胞增殖分化、调节细胞生长凋亡[1~3]。最近研究发现PHGPx在精子发生中发挥重要生物学功能,位于精子细胞及成熟精子上,且PHGPx基因内含子1a上存在着丰富的表达调控位点,与诸多生理机能有着密切联系[4],但目前硒蛋白PHGPx与睾丸硒元素、精子发生的调控机制还不清楚,本试验将构建山羊PHGPx基因干扰质粒,瞬时转染的生精细胞支持细胞共培养体系,用RT-PCR和 Western印迹方法检验沉默体外培养生精细胞PHGPx的表达,评价混合细胞PHGPx沉默效果,为进一步研究PHGPx在精子发生中作用及精子细胞硒蛋白代谢调控机制建立一良好的体外模型。
1 材料与方法
1.1 材料
经鉴定已传至6代山羊睾丸共培养的生精细胞由山西农业大学动物遗传育种与繁殖实验室提供。
1.2 试验方法
1.2.1 si RNA序列设计
PHGPx-si RNA模板寡核苷酸根据Gen Bank上的山羊PHGPx mRNA序列(Gen Bank No:GQ 302986),按照 Tuschl sh RNA 设计原则[9],利用Ambion公司在线siRNA设计软件(http://www.ambion.com/techlib/misc/si RNA_finder.ht ml)设计4条长度为21bp带发夹结构的比靶序列,分别位于150位、182位、214位和248位,4条特异性模板及阴性对照序列见表1。根据sh RNA表达载体p GPU6/GFP/Neo酶切位点的要求,送交上海吉玛生物技术公司合成带有Bbs I、BamH I酶切粘性末端和中央的7个碱基的发夹状序列。同时,按照Tuschl原则另设计一对阴性对照序列GTTCTCCGAACGTGTCACGT,经BLAST比对与现有基因文库中山羊源基因无同源性。

表1 设计siRNA序列信息Table 1 The sequence information of siRNA
1.2.2 si RNA干扰载体的构建
溶解并稀释相应的正、反寡核苷酸至终浓度为100μM;按照正反向si RNA寡居核苷酸模板各5.0μL、10×sh DNA Annealing Buff er 5.0μL和dd H2O 35μL共50μL的反应体系,95℃5 min、85℃5 min、75℃5 min、70℃5 min和4℃1 h的反应条件将PHGPx-si RNA寡核苷酸模板退火,退火处理后得到浓度为10μM的sh RNA模板。然后用限制性内切酶Bbs I、Ba m H I线性化的p GPU6/GFP/Neo载体与退火的 PHGPx-si RNA 双链寡核苷酸通过DNA连接酶连接,得到PHGPxsi RNA-p GPU6/GFP/Neo干扰重组质粒。再将10μL重组 PHGPx-si RNA-p GPU6/GFP/Neo质粒DNA转化到大肠杆菌感受态细菌,涂布与含Kana(40 g·L-1)的LB固体培养基上,37℃正置45 min待菌液至培养基完全吸收后倒置避光培养16 h以上,从转化培养的平板中挑取生长良好的单克隆菌落,接种于含有Kana(40 mg·L-1)的30 mL LB液体培养基中,37℃200 r·min-1摇床培养过夜,碱裂解法提取质粒DNA,所得质粒用Ba m H I和Pst I分别酶切鉴定。阳性重组载体应该可以被BamH I切开,而不能被Pst I切开。
1.2.3 PHGPx-si RNA-p GPU6/GFP/Neo 质 粒转染共培养生精细胞和支持细胞
取正常传至6~7代的共培养山羊睾丸生精细胞,0.25%胰酶消化后接种于覆有洁净载玻片的6孔培养盘内,接种密度为2~8×105·孔-1,加入2 mL·孔-1完全培养液继续培养24~48 h,使细胞生长状态稳定,细胞贴壁融合率80%~90%以上;转染前一天,吸除培养板中的旧培养液,使用无血清培养液洗涤细胞2次,加入2 mL Opti-MEM I Reduced Ser u m Mediu m继续培养细胞;吸除培养板中旧培养液,加入1.5 mL Opti-MEM I Reduced Seru m Mediu m,再按照500μL·孔-1加入转染复合物(10μL Lipof ectamine 2000 T M、4μg PHGPx-si RNA-p GPU6/GFP/Neo质粒和490μL Opti-MEM I Reduced Ser u m Mediu m),同时设置4次重复,轻轻振摇以混匀。4~6 h后,吸除培养液中止转染,更换完全培养液继续培养24~72 h观察转染效果。根据所构建干扰载体上绿色荧光蛋白(Green Fl uorescent Pr otein,GFP)表达后发绿色荧光的特点,在荧光显微镜下观察GFP的表达情况,计算质粒转染效率。
质粒转染效率=每高倍镜视野发荧光细胞数/同一视野下细胞总数×100%。
1.2.4 瞬时转染细胞PHGPx基因mRNA表达检测
转染后48 h细胞按照Trizol Reagent试剂盒操作说明提取组织的总RNA,DEPC处理水溶解,紫外分光光度计测定总RNA的浓度和纯度,调整浓度至1 g·L-1左右,用1%的琼脂糖凝胶分析总RNA的完整性,-80℃保存。按照SYBR Pri me-Script T M RT Reagent Kit试剂盒操作说明合成c DNA,用随机引物对总RNA进行反转录,产物-20℃保存。荧光定量PCR用PHGPx引物F:CAGGAGCCAGGGAGTAATG,R:TGAAAGT CCAGCCCAAGG(121bp);内参 GAPDH 引物F:TCCACGGCACAGTCAAGG,R:TCAGCACCAGCATCACCC(110bp);进行看家基因和目的基因的相对定量分析,其结果应用MX3000P软件(Stratagene公司)导出。
1.2.5 瞬时转染细胞PHGPx蛋白表达检测
转染后将6孔板内培养液吸除,使用预冷的PBS洗涤细胞2~3次,然后按0.1 mL·孔-1加入单去污裂解液(用前加PMSF抑制剂),充分吹打,冰浴放置1 h,转移至1.5 mL EP管。冰浴中超声间歇破碎细胞5次,20 s·次-1,避免起泡沫。4℃12 000 r·min-1离心10 min,保留上清,用 Nanodrop-1000核酸蛋白测定仪测定其蛋白浓度。然后将已知浓度的细胞蛋白样品加入5×上样缓冲液至终浓度为1×,在沸水中使蛋白变性7~10 min,即可点样进行SDS-PAGE电泳,电泳时间5~6 h,电压60~80 V,溴酚蓝刚跑出胶即可终止电泳进行转膜;转移结束后,将PVDF膜用TBS冲洗2~3次,移至装有封闭液的平皿,室温下封闭1 h;滤纸吸干PVDF膜上残留的封闭液后,用TBST 1∶100稀释PHGPx抗体,放置于杂交袋中37℃孵育2 h或者4℃过夜。一抗孵育完成后TBST洗膜2次,每次10~15 min,再用TBS洗膜1次,10 min,按1∶1000比例稀释二抗,室温下孵育1 h,二抗孵育完成后TBST洗膜2次,每次10~15 min,再用TBS洗膜1次,10 min,即可进行显色反应。化学发光、显影定影参照博士德公司ECM化学发光试剂盒操作,X-光胶片可见光下成像观察印迹条带,保存图像。
1.3 统计分析
采用-ΔΔCT方法获得PHGPx mRNA的相对表达量和使用I mage Pr o Pl us 6.0图像分析软件分析内参蛋白β-actin和PHGPx蛋白平均光密度值,数值利用SPSS软件进行统计学分析。
2 结果与分析
2.1 PHGPx-si RNA表达载体的构建
重组载体酶切鉴定结果见图1。p GPU6/GFP/Neo具有Pst I酶切位点,当插入目的基因片段后,Pst I酶切位点被破坏,而Bam H I酶切位点保留,故重组质粒经Pst I和BamH I酶切鉴定,阳性克隆不被Pst I切开,电泳结果显示多重构象,经BamH I线性化,阳性克隆为单一线性化条带。重组载体测序结果见图2,所设计的si RNA片段成功连入p GPU6/GFP/Neo载体,可进行后续试验。
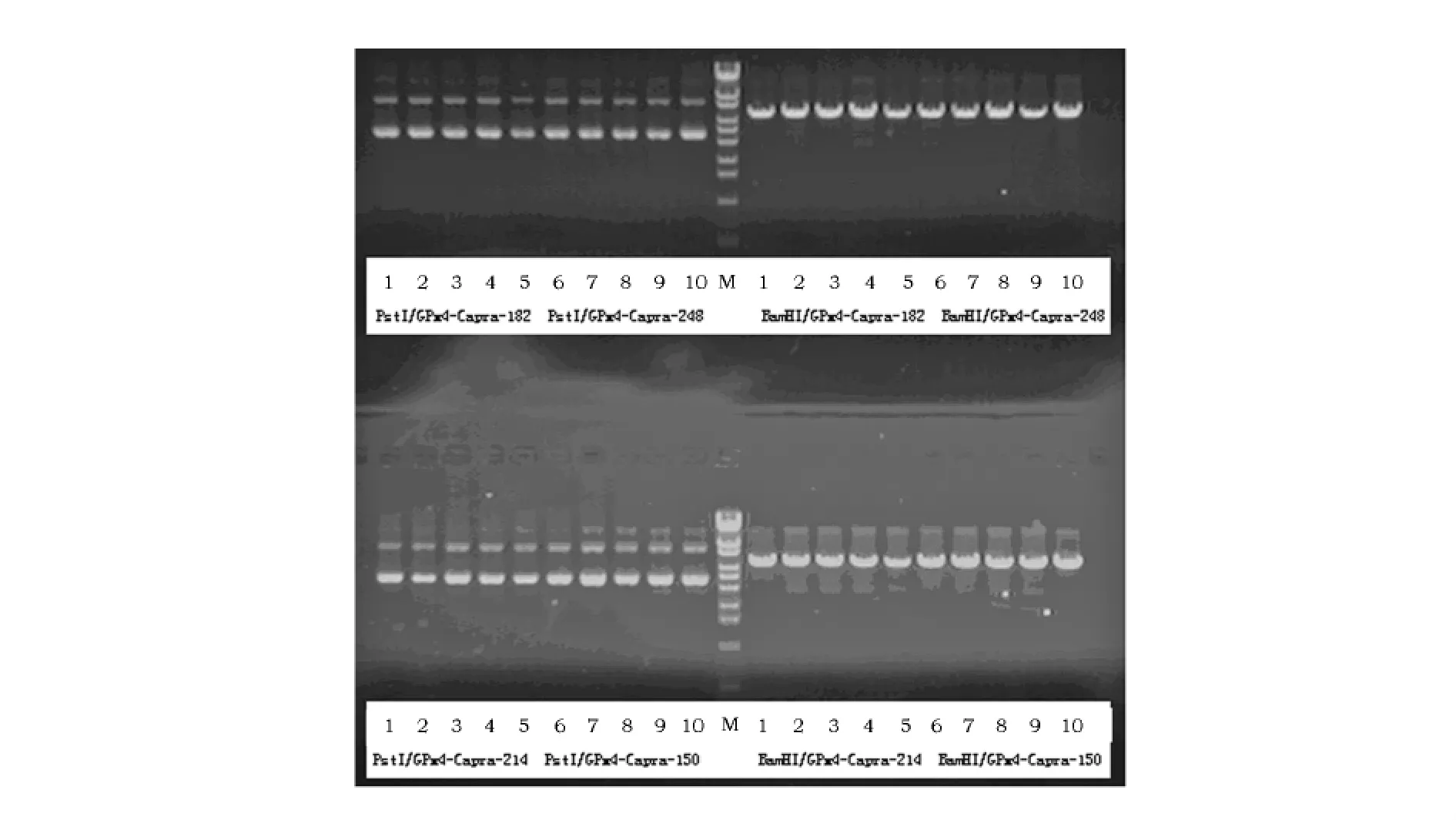
图1 重组载体的酶切鉴定Fig.1 Identification of recombinant plas mids with enzy me digestion
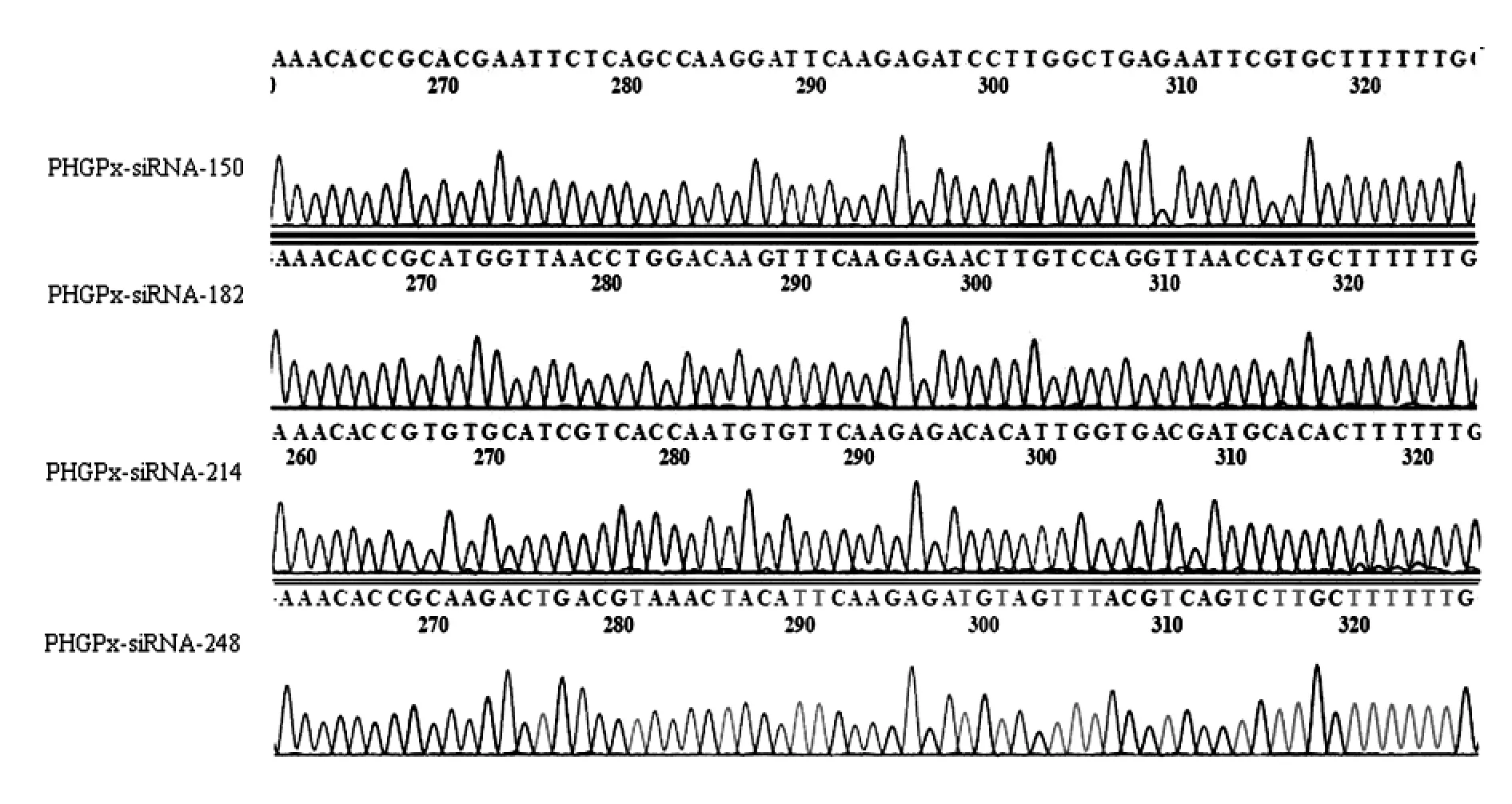
图2 PHGPx-si RNA序列图Fig.2 Sequence of PHGPx-si RNA
2.2 瞬时转染后共培养睾丸生精细胞荧光信号检测
带有绿色荧光蛋白标记的重组si RNA表达载体经常规瞬时转染步骤转入体外共培养山羊睾丸生精细胞后,通过荧光显微镜检测绿色荧光蛋白表达,结果如图3所示。24~48 h内,绿色荧光蛋白能够正常表达,荧光显微镜下检测到较强的绿色荧光信号,表明转染成功,在转染后24 h可进行转染效率评价。
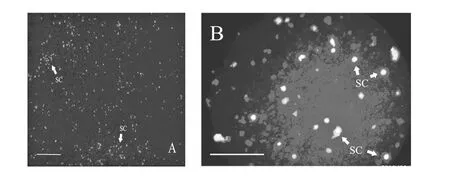
图3 pGPU6/GFP/Neo载体转染共培养生精细胞荧光结果Fig.3 The expression of p GPU6/GFP/Neo vector green fluorescent protein in coculturesper matogenic cells after 24 htrans fection
2.3 瞬时转染后共培养睾丸细胞中PHGPx mR NA表达
Real-time PCR检测各组共培养生精细胞中PHGPx mRNA表达水平见图4。PHGPx和GAPDH根据Real-time PCR结果显示,阴性对照、只加转染试剂组和空白对照细胞中PHGPx mRNA表达差异不显著(P>0.05),转染组与对照组相比,除182位点的重组质粒对体外共培养生精细胞PHGPx mRNA表达水平没有干扰作用外,150、214和248位点的重组质粒极显著的抑制了生精细胞PHGPx mRNA的表达(P<0.01)。
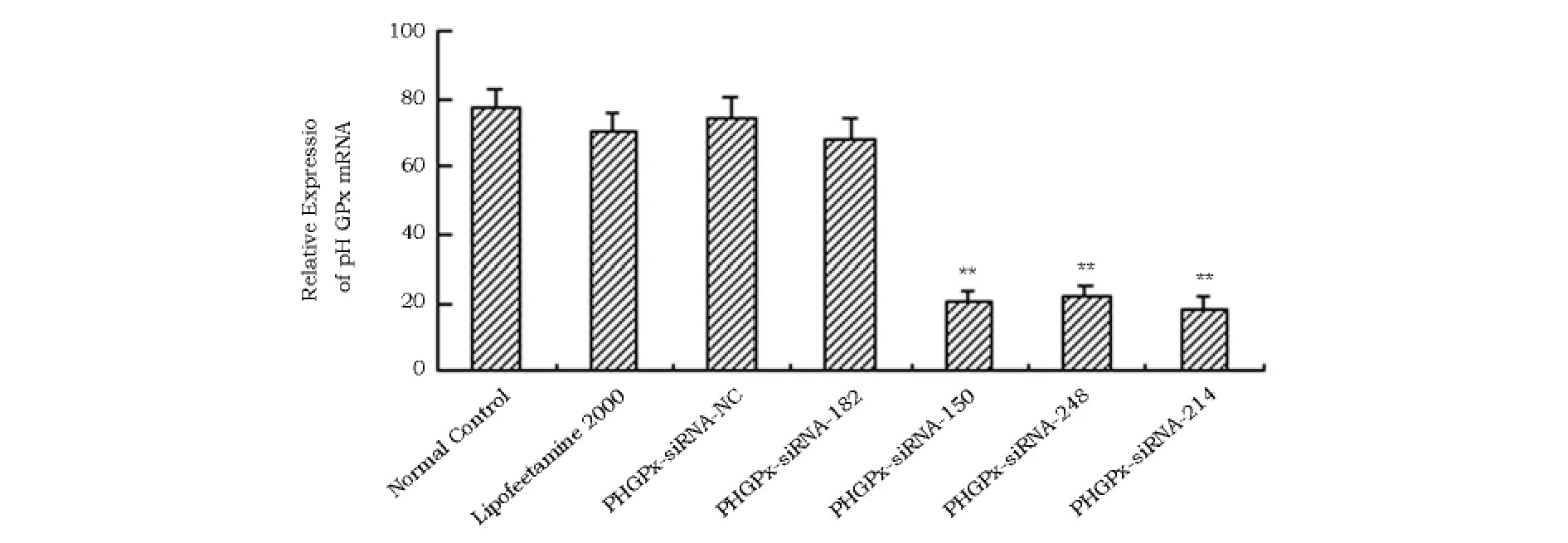
图4 Real-time PCR检测瞬时转染48 h后共培养生精细胞PHGPx mRNA表达Fig.4 The expression of coculture spermatogenic cells PHGPx mRNA after 48 htransfection
2.4 瞬时转染后共培养睾丸细胞中PHGPx蛋白表达
转染72 h后,以β-actin为内参,Wester nblotting检测转染组和对照组PHGPx蛋白表达水平。结果如图5 A所示,根据平均光密度值,182位点PHGPx-si RNA重组质粒对体外共培养山羊生精细胞PHGPx蛋白表达干扰效果较差,150和248位点设计的重组质粒对PHGPx干扰效果显著(P<0.05),214位点设计PHGPx-si RNA重组质粒对共培养生精细胞PHGPx蛋白表达干扰效果极显著,表达水平最低(P<0.01),图5 B为转染组生精细胞内参β-actin和 PHGPx蛋白 Western-blotting结果,与Real-time PCR检测结果基本一致。
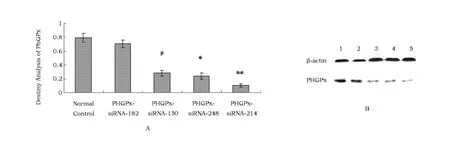
图5 Western-blotting检测瞬时转染72 h后共培养生精细胞PHGPx蛋白表达Fig.5 The expression of coculture sper matogenic cells PHGPx protein after 72 htransfection
3 讨论
RNA干扰(RNA interference,RNAi)目前已被广泛用于功能基因组和基因治疗的研究,成为最受欢迎的技术之一。而以质粒为基础的RNA干扰技术因其成本低廉、操作方便、抑制效率高以及能够筛选稳定抑制靶基因表达的细胞克隆等诸多优点受到越来越多研究者的青睐[5,6]。本研究通过建立以质粒为基础的RNA干扰技术,研究重组质粒对山羊共培养生精细胞PHGPx表达干扰效果。
3.1 转染效果的检测
RNAi效果的检测可以通过形态观察进行,当生物体内的靶基因被沉默后,如果该基因是关键基因,生物体会在形态或行为上表现出异常或者死亡,但如果在形态或行为上看不出明显的变化,则需要从分子水平进行检测,如RT-PCR,或者是Wester n-bloting等从mRNA水平到蛋白水平的检测,而且分子水平检测更具有说服力[7]。由于RNAi仅针对靶向基因的mRNA而非蛋白,一般情况下,si RNA在18 h左右就可引起特定靶向基因mRNA的快速减少,但蛋白质的减少需要更长的时间,本研究在转染后24 h通过载体绿色荧光报告蛋白的表达初步检测到了转染效果,在转染48~72 h后才通过Real-time PCR和蛋白免疫印记技术对RNAi干扰效果进行测定,保证了检测结果的科学性和准确性。通过-ΔΔCT法分析转染组合对照组PHGPx mRNA的表达差异,结果显示182、214和248位点构建的重组质粒对共培养生精细胞PHGPx mRNA的表达有极显著的抑制效果,结合蛋白免疫印记检测转染后各组细胞PHGPx蛋白表达的结果看,182、248位点抑制效果不如214位点对PHGPx蛋白表达抑制效果显著,但在总体趋势上是基本一致的,这说明本试验所设计的PHGPx-si RNA-p GPU6/GFP/Neo重组质粒仅针对靶向基因PHGPx的mRNA而非蛋白,能在分子水平敲出PHGPx mRNA的表达,为了确保干扰效果的稳定,建议选用214位点的si RNA重组质粒进行后续的深入研究。
3.2 关于PHGPx的RNAi
I mai等人报道敲出PHGPx基因的小鼠会导致胚胎发育早期死亡[8],随后通过Cre-Lox P系统发现新的PHGPx基因敲除方法用来胚胎PHGPx的功能[9];圆精子PHGPx基因的敲除引起雄性小鼠不育[10]。目前通过PHGPx基因敲除模型的建立来研究PHGPx的功能存在着许多不容忽视的问题。最主要的是PHGPx在胚胎发育阶段起重要作用的基因,如果被敲除,可能导致胚胎发育的终止,即产生致死突变,传统的基因敲除无法满足此类研究的需要。通过PHGPx RNAi技术,能在体外培养的细胞达到基因敲除的效果,研究它的功能,而且从si RNA设计到重组质粒的提取,操作步骤简单,实验要求条件不高,可为后续试验的开展和深入研究提供指导。
4 结论
本研究设计了4对针对山羊PHGPx的si RNA,通过体外退火,成功将具有发夹结构sh RNA连入线性化p GPU6/GFP/Neo载体,PHGPx-si RNA-p GPU6/GFP/Neo重组质粒采用常规瞬时转染方法导入山羊睾丸体外共培养生精细胞,筛选出了3对有明显干扰效果sh RNA,共培养生精细胞PHGPx mRNA和蛋白的表达显著降低,为今后研究PHGPx作用调控机制提供实验基础。
[1]Conrad M,Schneider M,Seiler A,etal.Physiological role of phospholipid hydroperoxide gl utathione peroxidase in mammals[J].Biological chemistry,2007,388(10):1019-1025.
[2]Liang H,Remmen H V,Frohlich V,etal.Gpx4 protects mitochondrial ATP generation against oxidative damage[J].Biochemical Biophysical Research Communications,2007,356:893-898.
[3]Maiorino M,Scapin M,Ursini F,etal.Distinct promoters deter mine alternative transcription of gpx-4 into phospholipid hydroperoxide gl utathione peroxidase variants[J].J Biol Chem,2003,278(36):34286-34290.
[4]Kehr S,Malinouski M,Finney L,etal.X-ray fl uorescence microscopy reveals the role of seleniu m in sper matogenesis[J].Journal of Molecular Biology,2009,389(5):808-818.
[5]Kunath T,Gish G,Lickert H,etal.Transgenic RNA interference in ES cell-derived embryos recapitulates a genetic null phenotype[J].Nat Biotechnol,2003,21(5):559-561.
[6]Miyagishi M,Su mi moto H,Miyoshi H,etal.Opti mization of an si RNA-expression system with an i mproved hairpin and its significant suppressive effects in mammalian cells[J].J Gene Med,2004,6(7):715-723.
[7]Mc Manus M T,Sharp P A.Gene silencing in mammals by small interfering RNAs[J].Nat Rev Genet,2002,3(10):737-47.
[8]Imai H,Hirao F,Sakamoto T,etal.Early embryonic lethality caused by targeted disruption of the mouse PHGPx gene[J].Biochem Biophys Res Commun,2003,305:278-286.
[9]Imai H.New strategy of f unctional Analysis of PHGPx Knockout Mice Model Using Transgenic Rescue Method and Cre-Lox P System[J].J Clin Biochem Nutr,2010,46:1-13.
[10]Imai H,Hakkaku N,Iwamoto R.Depletion of Selenoprotein GPx4 in Sper matocytes Causes Male Infertility in Mice[J].J Bio Chem,2009,284(47):32522-32532.
