基于线粒体D-loop基因探讨花鳗鲡的群体遗传多样性及其种群进化历史
2012-10-23尹绍武
丁 旭, 齐 鑫, 尹绍武
(1. 海南大学 海洋学院, 海南 海口 570228; 2. 南京师范大学 生命科学学院, 江苏 南京 210046)
基于线粒体D-loop基因探讨花鳗鲡的群体遗传多样性及其种群进化历史
丁 旭1, 齐 鑫1, 尹绍武2
(1. 海南大学 海洋学院, 海南 海口 570228; 2. 南京师范大学 生命科学学院, 江苏 南京 210046)
通过测定花鳗鲡(Anguillia marmorata)海南群体(HN)和菲律宾群体(PH)共 19尾个体的线粒体D-loop基因的核苷酸序列(约1017 bp), 分析了花鳗鲡的种群遗传结构。结果表明: A、T、G、C 4种核苷酸的平均含量分别为39.8%、28.3%、12.4%、19.4%, A+T含量(68.1%)明显高于G+C含量(31.8%)。所测序列中存在73个变异位点, 共有18个单倍型。其中海南群体的单倍型多样度(Hd)、核苷酸多态性(Pi)、平均核苷酸差异数(k)分别为0.982、0.21577和219.655, 而菲律宾群体的单倍型多样度、核苷酸多态性、平均核苷酸差异数分别为 1.000、0.26728和 271.821, 两个群体之间平均遗传距离(P)为0.3203。结果表明 2个群体遗传变异较大, 菲律宾群体的遗传多样性较海南群体更加丰富。通过构建NJ分子系统树表明2个群体的亲缘关系较近。利用中性检验Tajima’s D (海南群体D=1.35345, P>0.01;菲律宾群体D=0.79220, P>0.01)和Fs(海南群体Fs=3.759; 菲律宾群体Fs=2.231)探讨其种群历史, 表明花鳗鲡群体进化过程种群数量较为稳定。
花鳗鲡(Anguillia marmorata); 海南和菲律宾群体; 线粒体D-loop基因; 遗传多样性; 种群进化历史
鳗鲡(Anguillia)属鱼类(日本鳗(Anguillia japonica)、欧洲鳗(A. Anguillia)、美洲鳗(A. rostrata)等)具有特殊的生活史——在海水中产卵, 在淡水中生长。鳗鲡的产卵场远离海岸线数千公里, 受精卵发育成柳叶鳗随洋流游至河口, 在淡水中生长并发育至性成熟, 再洄游至海洋的产卵场繁衍后代[1]。在进化背景下, 海洋中多数鱼类种群可视为随机交配群体[2-3]。鳗鲡广泛分布于温带到热带区域, 种群之间仍存在随机交配[4]。但是, 花鳗鲡(A. marmorata)种群却具有特殊的种群结构和生活史[5]。花鳗鲡在分类学中属于硬骨鱼纲(Osteichthyes)、鳗鲡目(Anguilliformes)、鳗鲡科(Anguillidae)、鳗鲡属(Anguillia), 是一种广泛分布于西印度洋-南、北太平洋的热带和亚热带区域的典型鳗鱼。
关于鳗鲡种群结构的研究已有报道, Maes等[6]通过微卫星分析的方法发现欧洲鳗、美洲鳗、日本鳗和花鳗鲡具有高度的保守性。本实验室通过研究线粒体细胞色素 b, 认为花鳗鲡与日本鳗鲡较为相似, 亲缘关系较近, 且比欧洲鳗的进化程度更高;并认为花鳗鲡的夏威夷群体与日本群体间的地理差异较小, 但与海南群体的地理差异较大[7]。通过分析微卫星位点, 花鳗鲡的中国群体与澳洲群体具有较高的多样性指数[8]。Ishikawa[9]从遗传学角度把 花鳗鲡种群分为北太平洋、南太平洋和印度洋3个亚群, 其分布与现代海洋的洋流系统和水体结构相一致。在这些种群中仅仅发现了一个产卵场——位于北太平洋西部(菲律宾南部、斯里兰卡东部、巴布亚新几内亚和关岛西部之间)的深海海沟中[10-11]。Aoyama等[12-13]发现花鳗鲡比温带鳗鲡的产卵周期更长、生长速度更快、洄游路径更短。这表明花鳗鲡的种群结构比温带鳗鲡更加多样。因此,研究花鳗鲡独特的生理特点和种群结构, 能够完善花鳗鲡基因型多样性的证据, 为研究鳗鲡的起源、探索其产卵场位置、研究其洄游的生态学机制奠定基础。
基因存储了生物在进化过程中重要的遗传信息。而线粒体基因(mtDNA)是母系遗传, 其结构简单、进化速度快, 是研究种间和种内遗传多样性的重要的分子标记[14]。D-loop是线粒体DNA主要的非编码区, 它位于转运脯氨酸和苯丙氨酸的tRNA序列之间。由于受选择压力小, D-loop区片段在进化过程中积累了较多变异, 且比其他线粒体控制区进化的更快。D-loop区的 5′端显示了硬骨鱼类之间高水平的核苷酸替换, 有利于研究种内变异[15]。因此可以利用线粒体基因D-loop控制区研究花鳗鲡群体内部的遗传结构、亲缘关系。
本研究利用线粒体DNA的D-loop控制区首次比较分析了海南群体和菲律宾群体花鳗鲡的遗传多样性和系统进化关系。为了解花鳗鲡群体的遗传多样性和种群进化历史提供更多的证据。
1 材料与方法
1.1 样本采集与总基因组DNA的提取
2010年5月采集花鳗鲡玻璃鳗样本共19尾, 取自中国海南海域(11尾)和菲律宾南部海域(8尾), 为玻璃鳗时期样本, 体质量在123~465 g, 分别记为海南群体和菲律宾群体。采用常规“酚/氯仿”抽提法[16]从肌肉中提取花鳗鲡总DNA, 经乙醇纯化、重溶后, 用1.5%琼脂糖凝胶电泳结合 EB染色检测所提取的DNA质量, 用仪器Eppendorf biophotomete (Eppendorf AG 22331 Hamburg) 测定DNA浓度。将提取的DNA于-20℃保存备用。
1.2 PCR扩增和序列测定
根据GenBank中花鳗鲡mtDNA的D-loop基因序列片段(登录号: NC006540), 用Primer premier 5.0软件设计特异性引物。引物序列为 D-loop F: 5′-CGAGTAGAACCGTAGAAGTCA-3′和 D-loop R:5′-TCCATCCTCAACTCCCGAAG-3′。由上海生工生物工程技术服务有限公司合成。PCR反应总体积为20 μL, 其中 200 μmoL/L dNTP、2.5 μL 10×buffer、2 μmoL/L MgCl2、0.4 μmoL/L 引物(各 0.2 μmoL/L)、2 U TagDNA聚合酶、20 ng DNA模板, 并用超纯水补充体积。PCR反应程序为: 94℃预变性5min; 94℃变性1 min, 54℃退火30 s, 72℃延伸1 min20 s, 共35个循环; 最后72℃延伸10 min。PCR产物用1%琼脂糖凝胶电泳, 凝胶成像系统观察, 对扩增的片段进行确认。PCR产物用OMEGA公司胶回收试剂盒纯化后, 由上海 Invitroge公司进行测序反应。为保证序列的准确性, 序列经过正反两次重复测定。
1.3 D-loop分析
采用Contig-Express软件对正、反向序列进行重叠区拼接, 除去多余的碱基片段。用BioEdit软件分析碱基含量。采用DNAsp4.0软件对多态位点数、单倍型数、转换与颠换数、核苷酸多样性指数(Pi)、单倍型多样度(Hd)、平均核苷酸差异数(k)、平均遗传距离(P)等遗传多样性参数进行计算。并利用中性检验研究花鳗鲡的种群历史。用 Mega5.0软件进行遗传距离计算和聚类分析。系统树采用NJ模型进行构建, 并用并采用 bootstrap(重复次数 1000)检验聚类树各分支置信度。
2 结果分析
2.1 群体序列多样性
本研究检测的19个样本中, 经校对得到长度在1017~1020 bp的同源序列。A、T、G、C碱基平均含量分别为 39.8%、28.3%、12.4%、19.4%。其中A+T含量(68.1%)明显高于 G+C含量(31.8%)。所有序列的共检测到73个核苷酸变异位点, 共存在18种单倍型(HN6和HN10共享同一个单倍型)。所有突变位点中存在4个缺失位点、4个颠换位点以及63个转换位点。
2.1.1 海南群体序列多样性
经测序得到11个海南群体的序列, 序列长度在1018~1020 bp, 平均长度为1019.18 bp。内部共检测到 56个核苷酸位点变异, 占全序列的 5.49%。其中缺失位点4个, 2个位点发生颠换, 48个位点发生转换, 平均转颠换比为 24.0, 碱基替换与插入或缺失的比例为8.33。这些位点分属于10个单倍型。
2.1.2 菲律宾群体序列多样性
对菲律宾群体 8个样本进行测序, 发现其序列长度在 1017~1020bp, 平均长度 1018.75bp。内部共产生碱基位点变异 48个, 占全序列的 9.93%。缺失位点1个, 3个颠换位点, 42个转换位点。平均转颠换比为 14.0, 碱基替换与插入或者缺失的比例为15.0。这些位点分属于8个单倍型。
综合 2个群体的分析结果, 发现它们的碱基替换位点明显大于插入或缺失位点, 碱基替换中转换又大于颠换。这完全符合线粒体基因组的进化规律。碱基转换与颠换的比例是多重替换程度的一个标准, 可以衡量序列间自遗传分化以来各个位点发生替换的饱和程度[17]。本研究中, 较高的平均转颠换比显示了群体间 D-loop基因的序列替换还没有达到饱和。
2.1.3 群体遗传多样性及遗传分化
用单倍型多样度(Hd)、核苷酸多态性(Pi)、平均核苷酸差异数(k)3个指标来衡量花鳗鲡 2个地理群体的遗传多样性(表1)。

表1 花鳗鲡2个群体的遗传多样性参数Tab. 1 Genetic diversities of two A. marmorata populations
结果显示, 海南群体的Hd为0.982低于菲律宾群体(Hd为1.000); 海南群体花鳗的Pi为0.21577也低于菲律宾群体(Pi为0.26728)。表明这两个群体具有较高的单倍型多样性(Hd>0.5), 和较高的遗传多样性(Pi>0.05), 且菲律宾花鳗的遗传多样性更加丰富。
2.2 群体内和群体间的遗传距离
根据 mtDNA D-loop区的序列, 用 Tamura-Nei的方法分析群体内部和群体间的遗传距离(表2)。结果表明, 18个单倍型之间遗传距离最大为0.7719, 最小为 0.0030; 总体平均遗传距离(P)为 0.3203。其中海南群体(HN)的单倍型遗传距离在 0.0059~0.6905,菲律宾群体(PH)的单倍型遗传距离在0.0059~0.5989,两个群体之间的遗传距离在 0.0030~0.7719。较大的遗传距离, 表明花鳗鲡的遗传多样性较高。遗传距离的最大值和最小值都出现在群体间, 可以推测两个地理群体之间的遗传分化尚不完全。

表2 基于花鳗鲡种群D-loop序列18个单倍型之间的遗传距离矩阵(左下角为遗传距离, 右上角为标准误)Tab. 2 Genetic distances (lower-left) and SE (what is se short for, should give the full name the first time it appears)(upper-right) among A. marmorata populations
2.3 系统发育结果
基于线粒体基因 D-loop控制区的全序列, 从GenBank上下载南半球和北半球赤道附近的 5个地理群体花鳗(斐济群体(FJ); 塔希提岛群体(TH); 关岛群体(GU); 苏拉威西岛群体(SU); 台湾群体(TW))的D-loop区序列(表3), 与本研究的2个地理群体(海南群体,菲律宾群体)的 18种单倍型一同进行系统发育分析。本研究使用 MEGA5.0软件中的 Kimura 2-parameter型构建花鳗鲡 NJ分子系统树(邻接树,图 1)。
结果显示所有的花鳗鲡群体可以明确分成两大支, 海南群体、菲律宾群体、台湾群体和苏拉威西岛群体聚为一支, 而余下的斐济群体、塔希提岛群体和关岛群体聚为另外一支。与各群体的地理分布结果一致。这表明海南群体和菲律宾群体的亲缘关系较近, 同属于北半球花鳗群体分支。
2.4 花鳗鲡种群历史分析
选择 Tajima[18]提出的D检验和 Fu[19]提出的Fu检验两种中性检验对花鳗鲡的种群历史进行分析。中性检验值 Tajima’sD(海南群体D=1.35345,P>0.01;菲律宾群体D=0.79220,P>0.01) 和Fs(海南群体Fs=3.759; 菲律宾群体Fs=2.231) 都为正, 且检验结果均不显著。暗示着海南和菲律宾的花鳗鲡群体在进化过程中可能经历了平衡选择的作用, 且群体大小维持稳定状态, 并未出现群体扩张或持续增长。
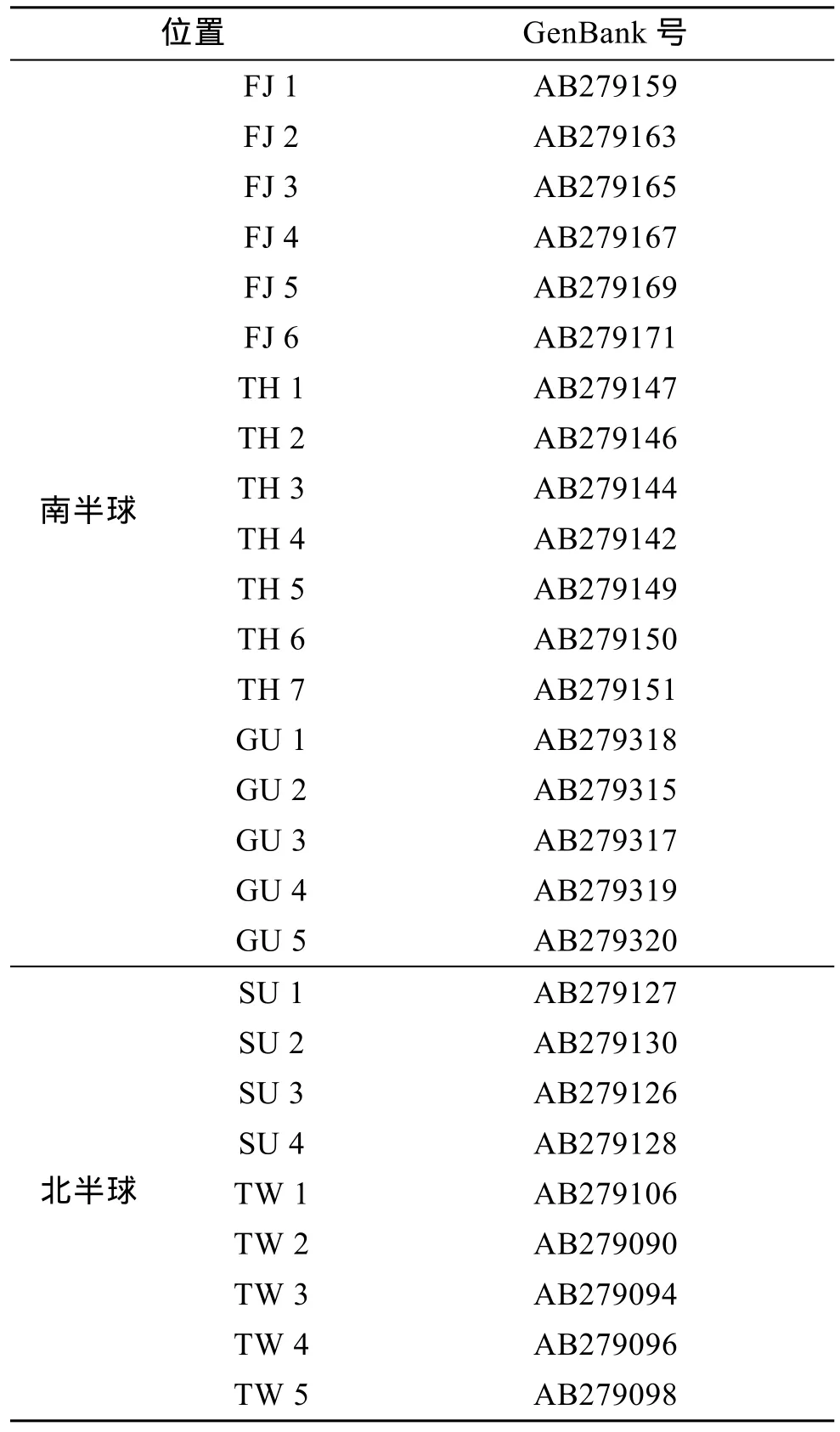
表3 花鳗鲡5个地理群的位置及GenBank号Tab. 3 Locations and the GenBank Accession numbers of the control regions in A. marmorata populations
3 讨论
3.1 花鳗鲡线粒体D-loop控制区的序列分析

图1 花鳗鲡mtDNA单倍型NJ分子系统树Fig. 1 NJ phylogenetic tree of mtDNA haplotypes in A.marmorata
本研究首次通过分析线粒体DNA的差异探讨了海南群体和菲律宾群体花鳗鲡的遗传多样性。本研究选取两个地理群体(海南群体和菲律宾群体)共 19尾花鳗鲡样本, 测序得到平均1019bp的碱基序列。分析两个花鳗鲡群体的线粒体D-loop基因序列表明,平均 A+T含量为 68.1%, 明显高于 G+C含量(31.8%)。这一结果与多数鱼类线粒体控制区的研究结果一致[20]。19个花鳗鲡样本共存在18种单倍型,海南群体内部有两个个体(HN4和HN10)共享一个单倍型。两群体之间均表现出各自独有的单倍型, 无共享单倍型存在。说明花鳗鲡各群体遗传多样性丰富,反映了花鳗鲡线粒体控制区进化速度快、序列变异大的特点。
两个地理群体内部的碱基突变结果显示, 海南花鳗鲡群体内部存在56个核苷酸位点变异, 而菲律宾群体则存在48个位点变异, 分别占群体之间总变异数的76.71%和65.75%。说明花鳗鲡的遗传变异主要存在于群体内部, 群体间的遗传变异较弱。在两个群体的线粒体控制区发现了共同的插入碱基, 暗示了它们之间存在一定程度基因交流。本研究中, 两个群体花鳗鲡的碱基转换位点明显多于颠换位点, 完全符合线粒体基因在进化过程中发生转换的频率通常远大于颠换频率且进化速度较快这一规律, 较高的平均转颠换比也表明花鳗鲡群体间D-loop基因序列替换尚未达到饱和, 推测花鳗鲡的这两个地理群体之间发生遗传分化的时间较短。
3.2 海南和菲律宾花鳗鲡群体 mtDNA D-loop控制区的遗传多样性
线粒体DNA的遗传多样性主要反映在单倍型平均遗传距离、单倍型多样度以及核苷酸多态性 3个方面[21]。本研究发现花鳗鲡的单倍型平均遗传距离为0.3203, 表明2个群体的变异极大, 遗传多样性极其丰富。遗传距离的最大值(0.7719)和最小值(0.0030)都出现在群体间, 可以推测 2个地理群体之间的遗传分化尚不完全。海南群体的单倍型多样度为0.982低于菲律宾群体(Hd为1.000), 表明这2个群体具有较高的单倍型多样性(Hd>0.5), 且菲律宾群体的单倍型更加丰富。Nei认为种群内部能够维持较高的单倍型多样性的原因可能在于较大的种群数量、环境的不均一性或者能够适应种群快速增长的生活习性[22]。海洋是一个大的开放型环境, 对海水鱼类而言,种群数量越大其维持遗传多样性的能力就越大[21]。而花鳗鲡广泛分布于西印度洋到南、北太平洋的热带和亚热带区域[2], 其种群数量极其庞大, 这可能是花鳗鲡维持其较高的遗传多样性的原因之一。
本研究发现海南群体花鳗的核苷酸多态性为0.21577低于菲律宾群体(Pi为0.26728)。显示了花鳗鲡较高的遗传多样性(Pi>0.05), 且菲律宾花鳗的遗传多样性更高。菲律宾群体较高的单倍型以及丰富的遗传多样性可能与其洄游路径有关。菲律宾花鳗的叶状幼体从产卵场随洋流游至菲律宾-印度尼西亚区域要经过较多个海峡, 再加上东南亚板块活跃的地理史[23], 导致菲律宾群体比海南群体花鳗鲡要经历更多的环境变化。此外, 近年来中国花鳗鲡种质资源下降(已被列为国家二级保护动物), 使得海南群体花鳗的遗传多样性降低。
不论是海南群体还是菲律宾群体, 它们的单倍型多样度都明显高于核苷酸多态性, 表明这 2个群体是由一个有效群体分化而来, 尽管在进化过程中积累了单倍型多态性, 但还不能使核苷酸序列多样化。结合Ishikawa[9]和 Katsuni[24]的研究成果, 可以推测海南群体花鳗和菲律宾群体都属于北太平洋群体。
3.3 花鳗鲡系统发育分析
线粒体基因进化速度比核基因快的多, 而D-loop区是线粒体基因中进化速度最快的区域, 能够提供更多的信息位点[25]。本研究通过邻位连接法构建的分子系统树, 发现各单倍型的节点支持率相对较高(图 1), 表明利用线粒体 DNA D-loop控制区可以有效的进行花鳗鲡系统发育学研究。图 1涉及的花鳗鲡主要来自北半球和南半球两大地理类群。NJ分子系统树显示南、北半球明显的分为两大支——南半球群体聚为一支, 北半球群体聚为另一支。尽管所算选取的花鳗鲡的地理位置都位于赤道附近, 但海南群体和菲律宾群体在分类上仍属于北半球群体。图 1也表明海南群体和菲律宾群体花鳗的亲缘关系非常近。花鳗鲡具有洄游路径短、产卵周期长、生长快速等特点[12-13]。Ishikawa[26]认为花鳗鲡存在多个产卵场, 南北半球不同的花鳗鲡群体可能来自于不同的产卵场。由于线粒体基因属于母系遗传, 由此作者推断花鳗鲡的海南群体和菲律宾群体同属于北太平洋群体, 他们都来自位于北太平洋西部的产卵场[10-11]。序列分析结果也显示花鳗鲡之间可能存在较多的基因交流。基因交流在某种程度上会阻碍群体间的遗传分化[27]。因此可以推测, 北半球的花鳗鲡群体发生不完全分化的时间较短。这与Katsuni[24]的关于花鳗鲡群体分化时间的结论一致, 也符合Maes[28]认为鳗鲡属鱼类起源较晚、演化速度较慢的观点。
3.4 花鳗鲡种群历史分析
种群历史主要包括种群扩增, 瓶颈效应, 奠基者效应, 群体缩减、分割和群体间的基因交流等。Kimura[29]在1968年提出了中性进化理论, 认为基因中的变异多数是中性的。遗传漂变、中性突变、群体大小的变化以及种群迁徙等随机事件是物种进化的主要动力。根据 Tajima[18]提出的中性检验方法(D检验), 若检验得到显著结果, 则偏离了中性模型,表明种群的遗传变异不单纯是由随机漂变造成的。而 Fu[19]提出的 Fu检验则是运用了种群遗传学溯祖理论, 对变异在不同分化时间上进行比较, 看是否符合中性检验。本研究显示两个地理群体的两种中性检验结果都为正值, 且没有显著差异(P>0.01), 表明花鳗鲡在群体进化过程中自然选择中的平衡选择起主要作用, 在进化史上并未出现群体扩张或持续增长, 群体大小保持稳定。
本研究发现这两个群体的花鳗鲡在进化过程中群体大小稳定、单倍型多样性高、遗传多样性高, 意味着具有较强的环境适应潜能以及生存、进化能力,对鳗鲡属鱼类种质资源的保护起到积极的作用。但从长远角度来看, 过度捕捞、水体环境污染会对其种质资源造成破坏, 拦河建坝以及水库、水电站的建立也会阻碍花鳗鲡的洄游路径, 使其遗传多样性降低。在中国, 花鳗鲡已列为国家二级保护动物。由于缺乏历史数据, 本研究尚不能确定花鳗鲡的遗传多样性是否由于过度捕捞等消极因素而受到影响。但是合理利用花鳗鲡资源, 制定可持续发展的保护措施,对于推动水产业的发展具有积极的意义。
[1] Tsukamoto K, Aoyama J, Miller M J. Migration,speciation, and the evolution of diadromy in anguillid eels[J]. Canadian Journal of Fisheries and Aquatic Sciences, 2002, 59: 1989-1998.
[2] Taylor E B. A review of local adaptations in Salmonidae,with particular reference to Pacific and Atlantic salmon[J]. Aquaculture, 1991, 98: 185-207.
[3] Adkison M D. Population differentiation in Pacific salmon:local adaptation, genetic drift, or the environment?[J]. Canadian Journal of Fisheries and Aquatic Sciences, 1995, 52: 131-145.
[4] Wirth T, Bernatchez L. Genetic evidence against panmixia in the European eel[J]. Nature, 2001, 409:1037-1040.
[5] Ege V. A revision of the genus Anguilla Shaw:a systematic, phylogenetic and geographical study[J]. Dana Rep, 1939, 16: 1-256.
[6] Maes G E, Pujolarl J M, Raeymaekers1 J A M, et al.Microsatellite conservation and Bayesian individual assignment in four Anguilla species[J].Molecular Ecology Progress Series, 2006, 319: 251-261.
[7] 齐兴柱, 尹绍武, 娄甜甜, 等. 海南产花鳗鲡细胞色素 b基因的克隆及序列分析[J]. 海南大学学报(自然科学版,) 2007, 25(4): 397-401.
[8] Gong X L, Ren S J, Chen S Q. Highly polymorphic microsatellite loci from the giant mottled eel (Anguilla marmorata)[J]. Molecular Ecology Resources, 2009,9(6): 1544-1547.
[9] Ishikawa S, Tsukamoto K, Nishida M. Genetic evidence for multiple geographic populations of the giant mottled eel (Anguilla marmorata )in the Pacific and Indian Oceans[J]. Ichthyological Research, 2004, 51: 343-353.
[10] Aoyama J, Mochioka N, Otake T, et al. Distribution and dispersal of anguillid leptocephali in the western Pacific Ocean revealed by molecular analysis[J]. Marine Biology, 1999, 188: 193-200.
[11] Miller M J, Mochioka N, Otake T, et al. Evidence of a spawning area of Anguilla marmorata in the western North Pacific[J]. Marine Biology, 2002, 140: 809-814.
[12] Aoyama J, Wouthuyzen S, Miller M J. Short-distance spawning migration of tropical freshwater eels[J]. Biological Bulletin, 2003, 204: 104-108.
[13] Arai T, Aoyama J, Ishikawa S. Early life history of tropical Anguilla leptocephali in the western Pacific Ocean[J]. Marine Biology, 2001, 138: 887-895.
[14] Avise J C, Arnold J, Ball R M. Intraspecific phylogeography: the mitochondrial DNA brige between population genetics and systematics[J]. Ann Rev Ecol Syst, 1987, 18: 489-522.
[15] 彭士明, 施兆鸿, 陈超, 等. 根据 mtDNA D-loop序列分析东海银鲳群体遗传多样性[J]. 海洋科学, 2010,34(2): 28-32.
[16] Sambrook J, Fritsch E F, Maniatis T.Molecular cloning:a laboratory manual[M]. New York: Cold Spring Harbor Laboratory Press, 1989. 463-470.
[17] 吴琛, 宋大祥, 朱明生. 12SrRNA基因第三结构域序列分析探讨蜘蛛若干重要类群的亲缘关系[J]. 蛛形学报, 2002, 11(2): 65-73.
[18] Tajima F. Statistical methods for testing the neutral mutation hypothesis by DNA polymorphism[J]. Genetics, 1989, 123: 585-595.
[19] Fu Y X. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection [J]. Genetics, 1997, 147: 915-925.
[20] 刘焕章. 鱼类线粒体DNA控制区的结构与进化:鳑以鲏鱼类为例[J]. 自然科学进展, 2002, 12(3): 266-270.
[21] Neigel J E, Avise J C.Application of random walk model to geographic distributions of animal mitochondrial DNA variation [J]. Genetics, 1993, 135(4): 1209-1220.
[22] Nei M. Molecular evolutionary genetics[M].New York:Columbia University Press, 1987: 287-326.
[23] Hall R. Biogeography and Geological Evolution of SE Asia[M]. London: Geological Society of London Special Publication, 1998: 153-184.
[24] Katsuni T, Minegishi Y, Aoyama J. Multiple population structure of the giant mottled eel(Anguilla marmorata)[J]. Molecular Ecology, 2008, 17: 3109-3122.
[25] Matthew A S, Scott V E. Dynamics and phylogenetic implications of mtDNA control region sequences in new world jays(Aves:Corvidae)[J]. Molecular Evolution, 2000, 51: 97-109.
[26] Ishikawa S. Molecular study on the population structure of Anguilla marmorata[D]. Tokyo: University of Tokyo, 1998. 1-256.
[27] 黄原. 分子系统学——原理、方法及应用[M]. 北京:中国农业出版社, 1998: 372.
[28] Maes G E, Pujolar J M, Raeymaekers J A M, et al. Microsatellite conservation and Bayesian individual assignment in four Anguilla species[J]. Marine Ecology Progress Series, 2006, 319: 251-261.
[29] Kimura M. Evolutionary rate at the molecular level [J].Nature, 1968, 217: 624-626.
Genetic variation and population evolutionary history of the giant mottled eel (Anguilla marmorata) based on the mitochondrial D-loop gene
DING Xu1, QI Xin1, YIN Shao-wu2
(1. The Ocean College of Hainan University, Haikou 570228, China; 2. College of Life Sciences, Nanjing Normal University, Nanjing 210046, China)
Oct., 14, 2011
Anguilla marmorata; Hainan and the Philippines populations; mitochondrial D-loop gene; genetic diversity; population history
The population genetic structures of the giant mottled eel from Hainan and Philippines were investigated by sequencing the mitochondrial control region (D-loop) gene for the first time. The average contents of A, T, G and C in the control region were 39.8%, 28.3%, 12.4% and 19.4%, respectively. There were 73 polymorphisms sites from the sequenced samples revealing 18 haplotypes. By calculating the haplotype diversity (Hd), nucleotide diversity (Pi) and average number of pairwise nucleotide difference (k), the Philippines population exhibited higher level of variability (Hd=1.000, Pi=0.26728, k=271.821) than the Hainan population (Hd=0.982, Pi=0.21577, k=219.655 and the genetic distance (P) was 0.3203 between two populations. By constructing the molecular phylogenetic tree with the method of NJ, the two geographic populations of the giant mottled eel did not show significant genetic difference. Besides, neutrality tests indicated a possible stable population in the population history of the giant mottled eel.
S921
A
1000-3096(2012)05-0117-07
2011-10-14;
2011-12-17
“十一五” 国家科技支撑计划重点资助项目(2007BAD29B03); 江苏高校优势学科建设工程资助项目
丁旭(1987-), 女, 硕士研究生, 主要从事鱼类种质资源与遗传育种研究, E-mail: athena1468@yahoo.com.cn; 尹绍武, 通信作者, 教授, E-mail: yinshaowu@163.com
(本文编辑:谭雪静)
