抗痛风药非布索坦的合成工艺优化
2012-10-15姚刚,杨威
姚 刚,杨 威
(湖北科技学院药学院,湖北 咸宁437100)
非布索坦,化学名为2-[3-氰基-4-(2-甲基丙氧基)苯基]-4-甲基-5-噻唑甲酸,为目前世界上最新的黄嘌呤氧化还原酶抑制剂,其通过高度选择性地作用于该氧化酶,减少体内尿酸生成,降低尿酸浓度,从而有效治疗痛风疾病[1]。目前,痛风的发病率不断上升,并呈现出向年轻人蔓延的趋势,而国内尚无上市产品,因此,研究非布索坦的合成工艺意义重大。作者以2-(3-甲酰 基-4-异 丁 氧 基-苯 基 )-4-甲 基-噻 唑-5-甲 酸 乙 酯(化合物Ⅲ)为原料经醛肟脱水、水解制得目标化合物非布索坦(化合物Ⅰ),并对工艺进行优化。合成路线如下:

1 实验
1.1 试剂与仪器
氢氧化钠、盐酸、乙酸乙酯、氯化钠、丙酮、异丙醇,成都科龙化工试剂厂;无水乙醇、二氯甲烷、四氢呋喃、盐酸羟胺,重庆川东化工厂;无水硫酸镁、无水甲酸、二水甲酸钠,天津天大化学试剂厂;上述试剂均为分析纯。2-(3-甲酰基-4-异丁氧基-苯基)-4-甲基-噻唑-5-甲酸乙酯,上海诺特生物科技有限公司。
DLSB-10/30型低温冷却液循环泵,郑州长城科工贸有限公司;R201D-Ⅱ型减压旋转蒸发仪,上海申顺生物科技有限公司;EF-2型三用紫外仪,上海安亭电子仪器厂;HX501T型电子天平,慈溪天东衡器厂;U-300型高效液相色谱仪,上海戴安仪器有限公司。
1.2 方法
1.2.1 2-(3-甲酰基-4-异丁氧基-苯基)-4-甲基-噻唑-5-甲酸乙酯(Ⅲ)的精制
向反应瓶中加入化合物Ⅲ粗品20g、丙酮700mL,开启搅拌。水浴加热至(56±2)℃,回流溶解10min,趁热过滤,除去不溶物。将滤液转入另一洁净的反应瓶中,降温至(15±2)℃,保温析晶1h。用冰盐浴降温至(0±2)℃,保温析晶2h。抽滤,用丙酮洗涤烧瓶及滤饼,尽量抽干。将物料转入单口瓶中,减压旋转烘干。得化合物Ⅲ16.0g,收率80.0%。
1.2.2 2-(3-氰基-4-异丁氧基-苯基)-4-甲基-噻唑-5-甲酸乙酯(Ⅱ)的合成[2~4]
向反应瓶中加入化合物Ⅲ的精制品10g、无水甲酸120mL,室温搅拌溶解。溶解澄清后,依次加入盐酸羟胺4.0g、二水甲酸钠14.97g。升温至(80±2)℃,反应4h后取样送检,并降温至(25±2)℃,期间有晶体析出。用冰盐浴降温至(0±2)℃,保温析晶2h。抽滤,用纯化水洗涤烧瓶并泡洗滤饼,抽干。将物料转入单口瓶中,减压旋转烘干,得化合物Ⅱ9.52g,收率96.1%。
1.2.3 非布索坦粗品的合成[5]
向反应瓶中加入化合物Ⅱ20g、无水乙醇300mL,室温搅拌均匀。滴加5mol·L-1的氢氧化钠溶液17.42mL,10min左右滴加完毕。升温至(45±2)℃,反应1.0h。停止加热,取样送检,水浴降温析晶0.5h。冰水浴降温至0℃左右,析晶1.5h。抽滤,得白色固体,转移至另一洁净的烧瓶中,加入纯水300mL,搅拌溶解。加入乙酸乙酯600mL,搅拌均匀,并用冰水浴降温至0℃左右时,滴加5mol·L-1盐酸150mL,10min左右滴加完毕。用广泛pH试纸测pH值为3左右时,撤去冰浴,将溶液转移至分液漏斗中,静置分层。收集有机相,水相用乙酸乙酯300 mL提取,合并有机相。将有机相分段浓缩,45℃减压旋蒸得白色或类白色固体即化合物Ⅰ粗品17.0g,收率91.5%。
1.2.4 非布索坦的精制[6]
向反应瓶中加入非布索坦粗品20g、无水乙醇200mL,开启搅拌,水浴加热至55~62℃,搅拌溶解。溶解后加入活性炭0.2g,搅拌脱色20min。过滤,将滤液转移至另一洁净的反应瓶中。自然冷却降温析晶0.5h。用冰盐浴降温至0℃左右,保温析晶3h。抽滤,用无水乙醇洗涤烧瓶及滤饼,抽干。将物料转入单口瓶中,减压旋转烘干,得化合物Ⅰ精制品18g,纯度99%以上,收率90%,总收率63.31%。
2 结果与讨论
2.1 非布索坦精制品的HPLC分析(图1)

图1 非布索坦精制品的高效液相色谱Fig.1 The HPLC chromatogram of purified febuxostat
2.2 1 HNMR分析(图2)

图2 化合物Ⅱ(a)和化合物Ⅰ(b)的1 HNMR图谱Fig.2 The 1 HNMR spectra of compoundⅡ(a),compoundⅠ(b)
化合物Ⅱ:1HNMR(CDCl3),δ:1.084~1.101(d,6H,2CH3),1.374~1.409(dd,3H,CH3),2.173~2.223(m,1H,CH),2.764(s,3H,CH3),3.892~3.908(d,2H,CH2),4.331~4.384(s,2H,CH2),7.001~7.023(d,1H,Ph-H),8.074~8.101(d,1H,Ph-H),8.168~8.173(s,1H,Ph-H)。
化合物Ⅰ:1HNMR(CDCl3),δ:1.084~1.101(d,6H,2CH3),2.173~2.223(m,1H,CH),2.764(s,3H,CH3),3.892~3.908(d,2H,CH2),7.001~7.023(d,1H,Ph-H),8.074~8.101(d,1H,Ph-H),8.168~8.173(s,1H,Ph-H)。
2.3 合成工艺的优化
2.3.1 化合物Ⅲ的精制
2.3.1.1 溶剂的选择
外购化合物Ⅲ的HPLC分析结果见表1 。

表1 外购化合物Ⅲ的HPLC分析结果Tab.1 The HPLC analysis results of compoundⅢ bought
由表1 可知,外购化合物Ⅲ不纯,很有可能在下一步反应中带入较多未知杂质,导致终产品难以精制合格,需将其精制后投料。
用不同溶剂精制外购化合物Ⅲ,结果见表2 。
由表2 可知,异丙醇精制效果不好,无水乙醇和丙酮精制效果较好。考虑到溶剂的统一,用无水乙醇作为精制溶剂,但实验发现无水乙醇精制的化合物Ⅲ投料时化合物Ⅱ有较多极性小的杂质(HPLC分析),且不易精制;而用丙酮精制的化合物Ⅲ投料时化合物Ⅱ只有一个极性较小的杂质。因此,选择丙酮作为化合物Ⅲ精制用溶剂。
2.3.1.2 丙酮用量的确定
实验发现,丙酮用量为30BV时,化合物Ⅲ在回流状态下才能溶解,但抽滤时极易析晶,不利于操作,损失较大;丙酮用量为35BV时,抽滤时不易析晶,利于操作;丙酮用量为40BV时,抽滤时不易析晶,利于操作,但收率下降3%左右。综合考虑,选择丙酮用量为35BV。
2.3.1.3 化合物Ⅲ精制前后的实验结果
以HPLC监测化合物Ⅲ精制前后的反应情况,结果见表3 。

表3 化合物Ⅲ精制前后的实验结果Tab.3 The experimental results for compoundⅢ before and after refining
由表3 可知,用丙酮精制的化合物Ⅲ投料,产物纯度高,杂质少。
2.3.2 化合物Ⅱ的合成
2.3.2.1 盐酸羟胺用量的确定
以HPLC监测反应情况,实验发现,盐酸羟胺与化合物Ⅲ的摩尔比分别为3.6∶1、2∶1、1.5∶1时,6h反应液的峰面积依次为95.06%、98.19%、97.78%,粗 品 纯 度 依 次 为 96.84%、98.33%、98.09%。故选择盐酸羟胺与化合物Ⅲ的摩尔比为2∶1。
2.3.2.2 二水甲酸钠用量的确定
以HPLC监测反应情况,实验发现,二水甲酸钠与化合物Ⅲ的摩尔比分别为3∶1、5∶1、7∶1时,反应液的峰面积依次为 98.19%(6h)、98.29%(5h)、98.08%(4h),粗品纯度依次为98.33%、98.82%、98.29%。综合考虑反应时间、成本和产品质量,选择二水甲酸钠与化合物Ⅲ的摩尔比为5∶1。
2.3.3 化合物Ⅰ的合成
2.3.3.1 反应时间的确定
以HPLC监测不同反应时间的实验结果见表4。
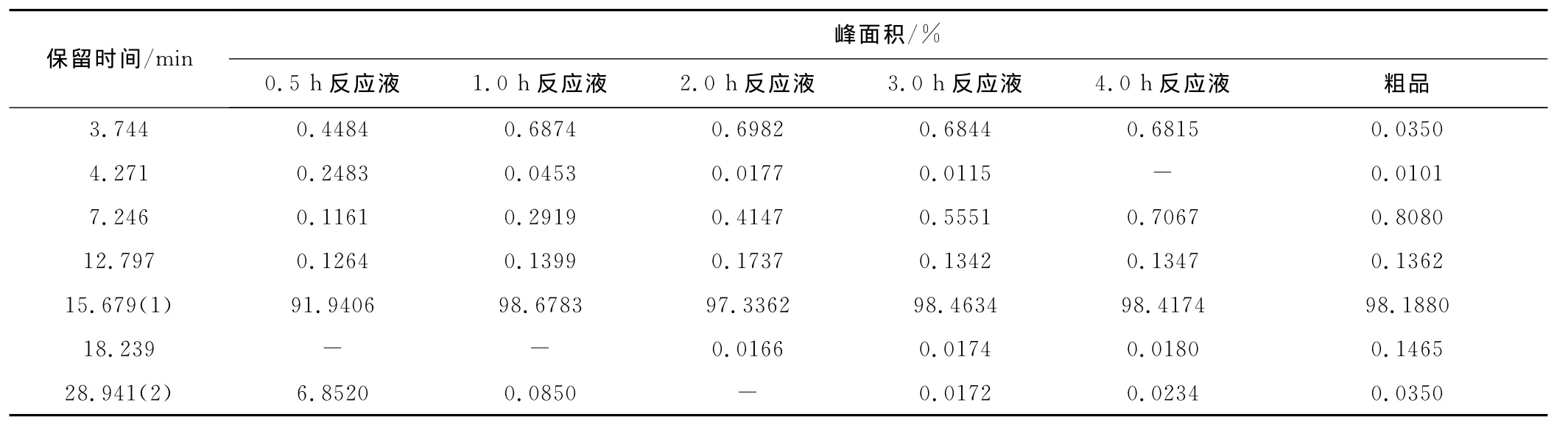
表4 不同反应时间的实验结果Tab.4 The experimental results at different reaction times
由表4可知,随着反应时间的延长,保留时间7min左右和18min左右的杂质含量增加。因此,确定最佳反应时间为1.0h。
2.3.3.2 盐酸浓度和pH 值的确定
反应时间为1.0h,保留时间18min左右的杂质是加盐酸后出现的,可以通过降低盐酸的浓度或用其它酸调pH值来调节。
以HPLC监测不同盐酸浓度和pH值条件下化合物Ⅰ粗品的纯度,结果见表5 。
由表5 可知,用5mol·L-1盐酸调pH值到3左右为最佳。
2.3.3.3 精制用溶剂的确定
以HPLC监测不同溶剂对化合物Ⅰ的精制效果,

表5 不同盐酸浓度和pH值条件下的化合物Ⅰ粗品纯度Tab.5 The purity of compoundⅠ crude at different hydrochloric acid concentrations and pH values
结果见表6。
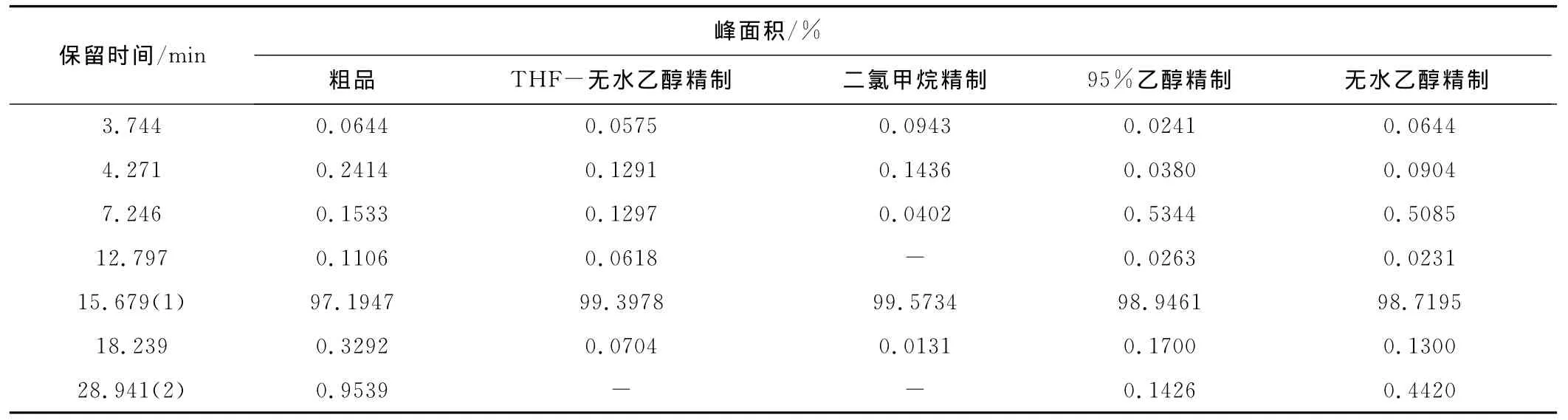
表6 不同溶剂对化合物Ⅰ的精制效果Tab.6 The refinery results of compoundⅠ by different solvents
由表6数据进一步计算,THF-无水乙醇、二氯甲烷、95%乙醇、无水乙醇精制的收率分别为34%、63%、75.7%、90%,其中精制效果较好的溶剂为无水乙醇。
3 结论
以2-(3-甲酰基-4-异丁氧基-苯基)-4-甲基-噻唑-5-甲酸乙酯为原料,用35BV丙酮精制后,与2倍物质的量的盐酸羟胺、5倍物质的量的二水甲酸钠,在甲酸作溶剂的条件下反应得2-(3-氰基-4-异丁氧基-苯基)-4-甲基-噻唑-5-甲酸乙酯;再与5mol·L-1NaOH 溶液反应1h,然后滴加5mol·L-1盐酸,pH值为3左右,低温条件下用乙酸乙酯萃取;最后用无水乙醇进行精制,纯度达99%以上,总收率达63.31%。优化后的工艺简单易控,非布索坦纯度及收率较高,达到药用标准,适合工业化生产。
[1]伍小云,胡艾希.非布索坦(Febuxostat)[J].中国药物化学杂志,2009,(4):319-320.
[2]孙运强,王玉玲,周辛波,等.非布索坦合成路线图解[J].中国医药工业杂志,2009,(12):954-956.
[3]Pascual E,Sivera F,Yasothan U,et al.Febuxostat[J].Nat Rev Drug Discov,2009,8(3):191-192.
[4]张嫒.抗痛风药Febuxostat[J].药学进展,2005,29(3):141-142.
[5]陈燕,罗宇,吕伟.非布索坦的合成[J].中国医药工业杂志,2009,40(1):1-5.
[6]郑凡,钱珊,杨莉,等.非布索坦的合成[J].中国医药工业杂志,2009,40(10):726-728.
