PCA方法在蛋白质亚细胞定位中应用
2012-09-28马军伟,史舵,顾宏,张杰
马 军 伟, 史 舵, 顾 宏, 张 杰
(1.大连理工大学 控制科学与工程学院,辽宁 大连 116024;2.山西省电力公司 电力通信中心,山西 太原 030001;3.安徽工业大学 数理学院,安徽 马鞍山 243002)
0 引 言
蛋白质功能的研究是蛋白质工程的重要环节,它对人们生活生产有着重要的意义,广泛应用于食品安全、新药研制、工业生产等领域,并在科学进步和社会发展中扮演推进剂的角色.如何更好地分析研究蛋白质的功能成为当前的主要问题.研究发现,蛋白质的功能与它的亚细胞定位密切相关,可以根据蛋白质的亚细胞位置来推断蛋白质的功能.传统蛋白质亚细胞定位预测方法主要是生物实验方法,如细胞分馏法、荧光显微法等.但是此类方法一般费用较高,而且比较费时[1].随着生物数据库中蛋白质序列数量的急剧膨胀,通过实验的方法获得蛋白质的亚细胞位置信息越来越不现实.因此,急需发展计算方法对蛋白质进行亚细胞定位预测.
序列编码技术是蛋白质亚细胞定位预测技术的基础,一般来说,按照基于氨基酸组成(amino acid composition,AAC)的编码方法,蛋白质将被表示为一个20维的特征向量,每一维对应一种氨基酸,以这种氨基酸在蛋白质序列中出现的频率为该维元素的值.然而,若用此种方法表示蛋白质,氨基酸之间的相对位置及序列长度都将遗失,从而造成表示上的固有局限性.为了解决此问题,Chou[2]提出了一个更高级的蛋白质表示模型——伪 氨 基 酸 组 成 (pseudo-amino acid composition,PseAAC),PseAAC 的应用显著提高了预测精度.PseAAC包含20+λ个成分,其中前20个成分按照AAC蛋白质序列方法编码,后λ个成分代表λ个不同级别的序列次序相关因子,这些离散的数列能近似地表示蛋白质的序列次序效应,从而提高了预测精度.近年来,PseAAC被广泛应用于生物预测模型[3、4].
一般来说,λ的值越大,包含的序列效益越大,但λ的值不能超过蛋白质的长度.若λ的值过大,则会造成如冗余和溢出之类的统计预测错误.因此,对于不同的训练数据集,应有不同的最优λ值.若数据集中最短蛋白质链的氨基酸残基数比较大,那么选择合适的λ值会比较耗时.本文在此情况下用主成分分析法提取主特征来解决这个问题.
1 理论分析
1.1 PseAAC
蛋白质的AAC特征表示由20个元素组成,分别代表20种不同氨基酸在蛋白质序列中出现的频率.伪氨基酸组成不但包含这些信息,还包含一些其他成分,通过这些成分近似反映蛋白质的序列顺序效应.
假设蛋白质链有L个氨基酸残基:

通过一系列序列顺序相关因子可以近似地反映序列次序效应,相关因子的定义如下:

其中λ<L.θ1称为第一级相关因子,反映蛋白质序列中相邻氨基酸相关性;θ2称为第二级相关因子,反映蛋白质序列中所有每间隔一个氨基酸的相关性;θλ称为第λ级相关因子,反映蛋白质序列中所有每间隔λ-1个氨基酸的相关性(图1).其相关函数Ci,j定义为

其中H1(Rj)、H2(Rj)和M(Rj)分别是Rj的疏水性值、亲水性值和侧链氨基酸质量.然后取代普通的基于氨基酸编码的方法,蛋白质X序列可以通过下式表示:

式中

其中fi表示蛋白质X中氨基酸的出现频率,θj表示蛋白质X第j级序列次序效应相关因子;w是序列次序效应的权重因子,在这里取w=0.05;λ表示此模型中采用的相关因子类型数量,且0≤λ<L.当λ=0时,说明模型中没有了能反映序列次序效应的相关因子,PseAAC便退化为普通的AAC模型;当0<λ<L,从式(3)、(4)中可以看出,前20个分量反映氨基酸组成效应,后λ个分量反映序列次序效应.由于λ个相关因子同序列次序效应紧密相关,也就是说λ越大,所包含的序列信息就越多,但是λ也有上限,必须小于蛋白质的氨基酸残基数目.此外,λ过大也有可能降低蛋白质的聚类性能,从而影响分类正确率.因此,对于一个给定的数据集,必存在一个最优的λ值.如果通过试验法获得,需要多次反复地试验才能找到最优值,将会特别费时费力.本文采用一种新方法来解决此问题.

图1 PseAAC示意图Fig.1 Schematic drawing of PseAAC
1.2 PCA
在PseAAC中前20个分量反映了蛋白质的组成信息,当其中一个量较大时同时会影响其他量的大小,后λ个分量反映了序列长度及次序效应,所以具有一定的相关性,从而说明数据在一定程度上有信息的重叠.主成分分析(PCA)采用一种降维的方式,找出几个综合因子来代表原来众多的特征,使这些综合因子尽可能地反映原来变量的信息,而且彼此之间互不相关,只研究样本特征中少数几个能最大程度保留原始特征变化方面信息的特征组合[5].这样也避免了求取最优λ值所带来的低效性.
假设所讨论问题有n个指标,可以看成n个随机变量,记为(X1X2…Xn),主成分分析的实质是线性组合这n个指标构成新指标

同时满足
(1)每一个主成分系数平方和为1,
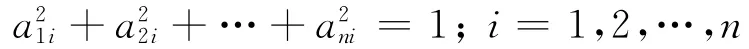
(2)主成分之间相互独立,

(3)主成分的方差依次递减,即重要性依次递减,

在求取时,可设X= (X1X2…Xn)T,新指标Y= (Y1Y2…Yn)T.则要在保持主成分之间相互独立且方差依次递减的原则下,找到变换矩阵U,有

可设X的协方差矩阵为根据方差的性质可知,ΣX是非负对称矩阵,所以必 存 在 正 交 矩 阵V, 使 得VTΣXV=中(λ1λ2…λn)是ΣX的特征向量,可以假设λ1≥λ2≥…≥λn,V正好是特征值对应的特征向量矩阵.可以证明V便是所要求的U,且各个主成分的方差var(Yi)=λi(i=1,2,…,n)[5].
所以,第i个主分量的方差贡献率可以定义为反映了样本数据的信息变化情况.前m(m<n)个主分量的累积方差贡献率定义为,可以选取前m个主分量使其累积方差贡献率达到一定的要求(如85%~95%),这样便可达到降低原始数据维数的目的.
2 实验准备
2.1 分类器
为了说明本文方法的有效性,选用两个比较常用的分类器:k近邻(k-NN)算法及反向传播网络(back-propagation network,BP network).
k-NN算法是一种比较简单成熟的分类算法,但其良好的分类性能并不弱于其他较复杂算法,已广泛应用于蛋白质二级结构预测及亚细胞定位.这里k取3,它被认为比1-NN有更好的可解释性[6],·2用来测量样本之间的相似度.
BP网络学习算法对于逼近实数值、离散值和向量值的目标函数有很强的健壮性,已被成功应用于人脸识别、视觉导航和生物信息学等领域.本文选用具有8个神经元的单隐藏层网络结构,基于变学习率的后向传播算法用来更新权值.最大训练次数设为300,同时为了防止过拟合,在每一次训练时都会估计泛化误差,若连续5次训练误差没有变化则训练会被提前终止[7].
2.2 数据集
本文选用2个不同的数据集来验证算法性能,分别进行PCA优化并观察优化之后的结果相比优化之前结果的改进程度.
第一个数据集由Chen等[8]构造,含有315个蛋白质序列(去掉两个已不用的蛋白质序列),具有6类亚细胞:细胞质蛋白质序列110个,细胞质膜蛋白质序列55个,线粒体蛋白质序列34个,分泌蛋白质序列17个,细胞核蛋白质序列52个,内质网蛋白质序列47个.第二个数据集由Gardy等[9]构造,已广泛应用于蛋白质亚细胞定位中.其含有541个蛋白质序列,具有4类亚细胞:细胞质蛋白质序列194个,细胞质膜蛋白质序列103个,细胞壁蛋白质序列61个,细胞外蛋白质序列183个.为方便使用,这两个数据库分别记作CH315和GA541.
2.3 精度评测
采用蛋白质亚细胞定位研究中常用的5折交叉验证法.具体做法如下:将数据集均分成5等份,选择其中4个子集作为训练集,然后选择第5个子集作为测试集,重复5次保证每一个子集都担任过测试集.实际上在神经网络实验中,用了5次5折交叉验证法来尽可能保证结果的合理性,这是因为梯度下降算法会收敛到相对于网络权值的局部极小值,而不是全局最小值.同时选用总体预测精度At作为评价指标,它被看作判断一个分类器好坏最重要的评价标准.定义如下:

其中P(i)表示第i类蛋白质序列被正确识别的数量,N是蛋白质序列的总数量,k是类别数量.
3 实验结果
不同的数据集中最短蛋白质序列长度是不同的,根据1.1节分析知λ必须小于最短序列长度,而且λ越大所包含的序列顺序效应越大.在数据集CH315中蛋白质序列长度最短为87,在GA541中最短为40.所以在使用PCA对数据集进行优化时,λ取最大值,分别为86和39.相应地,根据式(3),其输入向量维数分别是20+86=106和20+39=59.在实验中,累积方差贡献率设置为90%以提取关键主成分.
图2显示的是k-NN算法在CH315与GA541数据集中的预测精度与λ之间的关系.为了增加对比效应,PCA作用后的效果图已画出(虚线部分).可以看出预测精度随着λ取值不同在不断变化.在CH315数据集中,有3个最优λ值,分别是73、74和75,对应精度约为73.80%.在GA541数据集中,最优λ值是2,对应精度约为82.25%.经过PCA优化后,预测精度分别达到74.15%和82.46%,显然要高于最优λ值对应的精度.更重要的是,免去了求解最优值所带来的低效率.

图2 k-NN算法在两个数据集上的总体预测精度Fig.2 Total prediction accuracy using k-NN algorithm on the two datasets
图3 是BP神经网络在CH315与GA541数据集上的预测精度与λ关系图(虚线对应着PCA作用后的情况).不仅给出了每种情况下5次交叉验证的均值,同时画出了方差效果图.可以看出PCA作用后均值明显提升,同时方差变化范围明显减小,尤其在CH315数据集上表现更为突出,显示了PCA的优势.

图3 BP网络在两个数据集上的总体预测精度Fig.3 Total prediction accuracy using BP network on the two datasets
4 结果讨论
当λ=0时,PseAAC退化为AAC,从上述实验可以看出PseAAC确实要优于AAC.但是,对于不同的λ此模型会产生不同的预测精度,为了得到最优解,可以采用试验的方法逐一探求λ值,但是这种方法既繁琐又低效.实际上,为了解决这一问题,Chou等[10]提出了集成方法,通过将不同λ值的PseAAC融合成一个整体,集成了210个独立分类器,但是并没有指出基底分类器的数目对输出结果的影响.此外,集成如此大量的分类器难免产生较大的计算复杂度.本文采用主成分分析法,通过提取关键主特征来解决这一问题,试验结果显示此方法确实可以提高蛋白质亚细胞定位预测的准确度.
进一步,也尝试PCA与其他分类器的结合.作者构造了神经网络集成器定位亚细胞.在文献[11]中,601个蛋白质序列从革兰阴性杆菌库中提出,其中包括140个细胞质序列,74个细胞外序列,280个内膜序列,38个外膜序列,69个周质序列.最短序列长度为29,所以在PCA过程中λ设为28,90%的累积方差贡献率依然被用于提取关键主成分.表1显示了分类器的性能比较,同时,为了增加对比度,性能强大的PSORTb分类器预测结果也已给出.从表中可以看出,经过PCA优化后,各个类的精度都是最高的,总体预测精度达到82.0%,比PSORTb分类器和集成学习器各高16.6%和5.0%,说明PCA确实抓住了蛋白质样本的主特征.
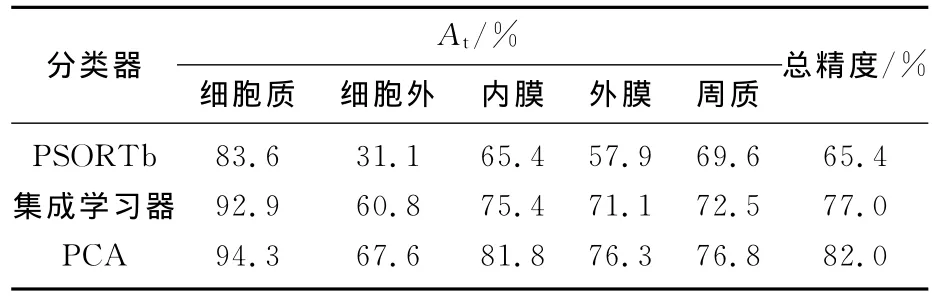
表1 3种不同方法的预测结果Tab.1 Predicted results by the three different methods
5 结 语
本文试验中用了90%的累积方差贡献率,当然这不是最优选择,在其他情况下也可以选择其他值,一般来说,大于等于85%的累积方差贡献率是合理的.另外,PCA只是一种线性变换,当数据高度复杂的时候效果并不明显,有时还有可能降低预测性能,下一步准备尝试kernel PCA(KPCA)、kernel independent component analysis(KICA)等非线性变换来替换主成分分析,对提升分类器的预测性能有一定帮助.
[1]SHEN Hong-bin, CHOU Kuo-chen. Predicting protein subnuclear location with optimized evidencetheoreticK-nearest classifier and pseudo amino acid composition [J]. Biochemical and Biophysical Research Communications,2005,337(3):752-756
[2]CHOU Kuo-chen. Prediction of protein cellular attributes using pseudo-amino acid composition [J].Proteins:Structure,Function,and Bioinformatics,2001,43(3):246-255
[3]DING Y S,ZHANG T L.Using Chou′s pseudo amino acid composition to predict subcellular localization of apoptosis proteins:an approach with immune genetic algorithm-based ensemble classifier[J].Pattern Recognition Letters,2008,29(13):1887-1892
[4]ZENG Y,GUO Y,XIAO R,etal.Using the augmented Chou′s pseudo amino acid composition for predicting protein submitochondria locations based on auto covariance approach [J].Journal of Theoretical Biology,2009,259(2):366-372
[5]李弼程,邵美珍,黄 洁.模式识别原理与应用[M].西安:西安电子科技大学出版社,2008
[6]VEENMAN C,REINDERS M.The nearest subclass classifier:A compromise between the nearest mean and nearest neighbor classifier [J]. IEEE Transactions on Pattern Analysis and Machine Intelligence,2005,27(9):1417-1429
[7]ZHOU Z H,WU J,TANG W.Ensembling neural networks:Many could be better than all [J].Artificial Intelligence,2002,137(1-2):239-263
[8]CHEN Y L,LI Q Z.Prediction of the subcellular location of apoptosis proteins [J].Journal of Theoretical Biology,2007,245(4):775-783
[9]GARDY J,LAIRD M,CHEN F,etal.PSORTb v.2.0:expanded prediction of bacterial protein subcellular localization and insights gained from comparative proteome analysis [J].Bioinformatics,2005,21(5):617-623
[10]CHOU Kuo-chen,SHEN Hong-bin.Large-scale predictions of gram-negative bacterial protein subcellular locations [J].Journal of Proteome Research,2006,5(12):3420-3428
[11]MA J W,LIU W Q,GU H.Predicting protein subcellular locations for gram negative bacteria using neural networks ensemble [C]// Proceedings of CIBCB′2009.Piscataway:IEEE,2009:114-120
