共沉淀法制备的MnOx-CeO2在含NO气氛中氧化碳烟的研究
2012-09-15付名利王克亮于润芃文贤儿张明黄碧纯吴军良叶代启3梁
付名利王克亮于润芃文贤儿张 明黄碧纯吴军良叶代启*,,3梁 红
(1华南理工大学 环境科学与工程学院,广州 510006)
(2华南理工大学工业聚集区污染控制与生态修复教育部重点实验室,广州 510006)
(3大气污染控制广东高校工程技术研究中心,广州 510006)
(4广州大学化学化工学院,广州 510006)
共沉淀法制备的MnOx-CeO2在含NO气氛中氧化碳烟的研究
付名利1,2王克亮1于润芃1文贤儿1张 明1黄碧纯1吴军良1叶代启*,1,3梁 红4
(1华南理工大学 环境科学与工程学院,广州 510006)
(2华南理工大学工业聚集区污染控制与生态修复教育部重点实验室,广州 510006)
(3大气污染控制广东高校工程技术研究中心,广州 510006)
(4广州大学化学化工学院,广州 510006)
用共沉淀法制备的复合氧化物MnOx-CeO2,其程序升温氧化(TPO)结果显示,1 000 mL·m-3NO和10%O2条件下MnOx-CeO2对应的碳烟起燃温度Ti为250~303℃,远低于无催化剂时的Ti(402℃)及CeO2的Ti(334℃);也低于无NO下MnOx-CeO2的Ti(346~360 ℃);与 MnOx的 Ti(290 ℃)相当,但 MnOx-CeO2的 Tm(413~441 ℃)仍比 MnOx的 Tm(441℃)稍低。明显地,NO 促进了碳烟的氧化,MnOx-CeO2比CeO2和MnOx的活性都要高。NO-TPD、FT-IR及原位DRIFTs表明,MnOx-CeO2表面对NO吸附能力强,更易促进NO氧化和NOx储存,从而有利于碳烟的氧化。可能的机理为,富氧条件下气相O2推动催化剂中氧物种(如超氧O2-,化学弱吸附氧O-与晶格氧O2-)的形成(含相互转化)与迁移,推进了NO或NO2-的氧化;储存的NOx在低温下生成硝酸根离子,在高温时则释放出高活性的NO2*和O-,促进碳烟氧化,其中间产物包括C-NO2复合物与C(O)复合物。
NO;MnOx-CeO2;碳烟氧化;催化剂;共沉淀法
0 引 言
柴油车排气中的NO可被氧化为NO2,NO2有比O2更强的氧化性,因此在某种程度上说NO可以促进碳烟的氧化[1-3]。MnOx与CeO2可形成复合氧化物MnOx-CeO2,其氧化还原能力更强,同样有利于碳烟的氧化[1,4-7]。NO在低温时以硝酸盐的形式储存于共沉淀法制备的MnOx-CeO2中,高温时硝酸盐分解释放出的NO2会促进碳烟氧化[1]。溶胶-凝胶法合成的MnOx-CeO2,因MnOx与CeO2的复合抑制了氧化物晶体的生长,提高了催化剂表面积和低温下的氧化还原性能,使催化剂氧化NO的能力增强,进而使NOx储存容量增大,对 1 000 mL·m-3NO+10%O2气氛下碳烟的氧化活性更好[3]。
前期工作中采用共沉淀法(CP)和柠檬酸配位燃烧法(CA)制备出Cu0.05-Ce0.95-O[8],形成的 Cu-Ce-O固溶体证实了Cu-Ce之间的相互作用,这决定了碳烟燃烧中的关键因素如催化剂的储氧容量(OSC,oxygen storage capacity),直接反映了其氧化还原能力,故Cu0.05-Ce0.95-O活性比CuO和CeO2要高。其中Cu0.05-Ce0.95-CA活性要优于Cu0.05-Ce0.95-CP。另有研究[9]表明:OSC并不是氧化碳烟催化剂的活性的唯一决定因素,活性氧物种如超氧(O2-)与晶格氧(O2-)作用同样也可能很大。
总之,MnOx-CeO2制备方法、前驱体、溶剂及Mn/Ce比例、焙烧温度等诸多条件[4,8,10]不同,催化剂获得的OSC、表面氧物种[3]或表面氧配合物[11]等活性因子数量或强度均有区别,加之反应气氛不同,催化剂活性与反应机理也不同。综合考虑MnOx与CeO2具有协同作用,共沉淀法可获得较好的固溶体结构,以及NO气氛有助于碳烟燃烧等几方面,本文采用共沉淀法制备MnOx-CeO2,以其醋酸盐为前驱体;并采用程序升温氧化(TPO)考察其催化氧化碳烟的活性,以及通过NO-TPD、FT-IR与DRIFTs研究催化剂表面活性物种特别是含氧、含氮物种及其迁移转化过程,探索碳烟氧化机理。
1 实验部分
1.1 催化剂的制备
实验采用共沉淀法[8]制备MnOx-CeO2复合氧化物,其基本原理是在含金属盐类的醋酸铈Mn(CH3COO)2·4H2O(上海晶纯试剂有限公司)、醋酸锰Ce(CH3COO)3·5H2O(天津福晨化学试剂厂)的水溶液中,以避免制备的催化剂中仍残留硝酸根(NO3-)对后续实验结果或表征分析的影响;加入沉淀剂氨水NH3·H2O(广州化学试剂厂),可以使 NH3在洗涤、干燥和煅烧过程中以气态的形式的形式挥发,降低残留在催化剂中的杂质;用JB50-D型增力电动搅拌机(上海标本模型厂)强力搅拌生成沉淀物,经静置、离心过滤后再依次用蒸馏水和无水乙醇 (成都联合化工试剂研究所)洗涤至中性。最后经100℃烘干3 h、550℃焙烧4 h,即得催化剂MnOx-CeO2。其中金属离子的物质的量比 nMn/(nMn+nCe)=0,0.1,0.2,0.3,0.4,0.5,1。
1.2 催化剂活性测试
程序升温氧化(TPO)催化剂活性评价[8]使用六气路气固相反应装置(天津鹏翔公司)。气氛为10%O2/He与1 000 mL·m-3NO+10%O2/He。实验所用模拟碳黑为德国Degussa公司的Printex®U碳黑,该碳黑在950℃时挥发份仅为5%,灰份含量低于0.02%,平均原生粒径则为25 nm,各项物化性质比较稳定,比实际碳烟更难燃烧,被国内外广泛采用模拟柴油车排气颗粒物。碳烟(30 mg)与催化剂(270 mg)简单混合(松接触状态,接近实际情况)。CO2检测采用配备了TCD和TDX-01的科创900A型气相色谱仪(上海科创公司)、N2000色谱数据工作站(浙江大学智能信息工程研究所)。以碳烟的起燃温度(Ti)作为催化剂活性的评价标准,Ti为当反应器出口尾气中 CO2浓度达到 5 000 mL·m-3时的温度[3];Tm为CO2浓度达到最大值时的峰温[3,12-14];Tc为碳烟燃尽时的温度[3,12-13]。
1.3 催化剂表征
NO升温脱附 (NO-TPD),采用美国麦克AutoChemⅡ2920全自动程序升温化学吸附仪。载气组成为5%NO/He混合气,载气流速为30 mL·min-1,催化剂用量为100 mg。先用He气在400℃下吹扫30 min,自然降温至60℃;切换至5%NO/He吸附90 min,再用He气吹扫30 min;最后待基线稳定后从60℃升温至500℃进行脱附实验,升温速率为 10 ℃·min-1。
FTIR:采用Nicolet 6700傅立叶变换红外光谱仪上对研磨成粉末的催化剂进行透射红外测试。测试时将催化剂与一定量的光谱纯KBr粉末混合研磨成微米级的粉体颗粒后压片(样品KBr=1wt%),压制成的薄片样品即可在设定条件下测试分析。分析条件:扫描范围 4 000~400 cm-1,扫描时间 64 s,分辨率 4 cm-1,检测器 DTGs,液氮制冷,采用 Omnic分析软件分析数据。
原位DRIFTs:通过Nicolet 6700型傅立叶变换红外光谱仪附带的Harrick漫反射原位池和高灵敏度MCT检测器进行分析。采样范围为4 000~650 cm-1,分辨率为4 cm-1,扫描次数为 256,进气流量为100 mL·min-1。催化剂粉末试样直接置于原位池的样品台内并压实,反应前于400℃用Ar吹扫0.5h除去表面杂质后,进行NO/(He)、NO+O2吸附与NO+O2气氛下反应催化剂的原位红外观测。
2 结果与讨论
2.1 MnOx-CeO2催化剂的活性测试(TPO)
为探讨CeO2和MnOx的相互作用,与NO对碳烟氧化的促进作用,分别在10%O2/He、1 000 mL·m-3NO+10%O2/He气氛中对MnOx-CeO2氧化碳烟进行了TPO测试,结果如图1所示。两种气氛下碳烟的燃烧温度Ti、Tm和Tc对比结果见图2。


如图1所示,10%O2/He气氛中,碳烟直接燃烧(无催化剂)的Ti为447℃,加入CeO2后降至381℃,下降了66℃;加入MnOx后降至360℃,下降幅度达87℃;当加入不同Mn/Ce比的催化剂时,Ti温度范围为340~360℃,下降幅度高达87~101℃。很明显,与CeO2和MnOx相比,MnOx-CeO2的活性更高(起燃温度Ti更低);且随着CeO2中MnOx含量的提高,碳烟的燃烧温度Ti、Tm和Tc首先保持较稳定,然后再下降。总的趋势为:MnOx含量增加,催化剂的活性提高(起燃温度 Ti降低)。其中 MnOx(0.4)-CeO2和MnOx(0.5)-CeO2活性相对较高。这反映了CeO2和MnOx之间存在相互作用,且与Mn/Ce比例相关[4]。
1 000 mL·m-3NO+10%O2/He气氛中,碳烟直接氧化(无催化剂)的Ti为402℃(低于10%O2/He气氛中 Ti,447 ℃),加入 CeO2后降至 334℃,下降幅度为68℃;加入MnOx后降至290℃,下降了112℃;当加入不同Mn/Ce比的催化剂时,Ti温度范围为250~303℃,下降幅度高达99℃~152℃。很明显,NO的存在促进了碳烟的氧化,MnOx-CeO2的活性也优于CeO2和MnOx。随着CeO2中MnOx含量的提高,碳烟的燃烧温度Ti、Tm和Tc先略有升高,然后再下降,总的趋势为:MnOx含量增加,催化剂的活性也提高。其中MnOx(0.5)-CeO2活性相对较高。这再次证明了CeO2和MnOx之间存在相互作用,但MnOx含量增加与催化剂的活性提高并无明显线性关系[4]。
如图2所示,相比于10%O2/He气氛,1 000 mL·m-3NO+10%O2/He中,同种催化剂氧化碳烟的Ti降低了47℃~98℃,Tm和Tc则分别降低了84℃~100℃和36℃~91℃。其中活性最好的是MnOx(0.5)-CeO2,起燃温度Ti降至250℃,且在实际柴油车排放温度范围内[15]。
综上,反应气氛中引入NO的确促进了碳烟的燃烧,但仍需要在NO气氛下进一步考察CeO2和MnOx之间的相互作用,拟重点对MnOx-CeO2的表面物种进行研究。
2.2NO-TPD
上述活性测试中发现了CeO2和MnOx之间的相互作用,复合氧化物中MnOx(0.4)-CeO2和MnOx(0.5)-CeO2活性相对较高;同时也发现了NO的存在促进了碳烟的燃烧。故需再对MnOx-CeO2进行NOTPD测试,考察其对NOx的吸附脱附性能,以进一步探究NO促进催化剂对碳烟的氧化,结果如图3所示。
由图3可见,各催化剂在90℃左右都出现一个明显的低温脱附峰。明显地,无论是低温脱附峰面积,还是脱附峰总面积,复合氧化物MnOx-CeO2都大于CeO2和MnOx,峰面积由高到低排序为MnOx(0.5)-CeO2> MnOx(0.3)-CeO2> MnOx(0.2)-CeO2> MnOx(0.1)-CeO2> MnOx(0.4)-CeO2> CeO2> MnOx,这与碳烟燃烧的燃烧完全温度Tc由低到高(对应催化剂活性实际由高到低)的顺序即 MnOx(0.5)-CeO2<MnOx(0.3)-CeO2≈MnOx(0.2)-CeO2<MnOx(0.1)-CeO2≈MnOx(0.4)-CeO2<CeO2<MnOx较为一致(见图 1(b)和图 2(b))。低温下NO的脱附峰面积和脱附峰总面积正相关于1 000 mL·m-3NO+10%O2气氛中催化剂对碳烟的氧化活性潜力,即脱附峰面积越大,越有利于碳烟的氧化[1]。
在270~300℃左右各催化剂则出现高温脱附峰,复合氧化物MnOx-CeO2中随着Mn含量的增多,高温峰峰宽逐渐增大,且主峰对应的温度逐渐向低温方向靠近,这与各催化剂活性差异情况基本一致。

2.3 FTIR
据图3分析结果,可以初步认为催化剂对NO吸附脱附性能越强,其活性便越高。因此,有必要进一步研究催化剂对NO的吸附、NO储存形式及其转化途径的影响因素,重点检测和分析催化剂表面含氧物种(如 O2-、O-与 O2-等)[16]、含氮物种等。
对NO-TPD后的催化剂进行了FTIR测试,结果如图4所示。据图4,各催化剂在1 380 cm-1处有明显的吸附峰,归属于离子NO3-的伸缩振动[3,17-19],即无气相O2参与时,由NO和催化剂体相中的氧物种,如晶格氧O2-[2,20]结合而成。其中MnOx-CeO2复合氧化物上的NO3-(硝酸盐)吸附峰强度稍大于单一氧化物CeO2和MnOx,这说明NO以硝酸盐的形式储存在催化剂表面。在没有气相O2的情况下,催化剂活性氧物种(O2-、O-与O2-)[16]在NO物种形态转化过程中起着关键作用,如晶格氧O2-可将NO氧化为NO2[9,19-20];相比于单一氧化物 CeO2和 MnOx,MnOx-CeO2复合氧化物活性氧物种更为丰富,其吸附转化NO最后生成并存储的NO3-物种更多,这与NO-TPD的结果一致。引入NO后,碳烟起燃温度范围为250~303℃,这与MnOx-CeO2的NO高温脱附峰温度范围(270~300℃)较好地吻合,进一步证实NO+O2气氛中NO的吸附和NOx的脱附增强了MnOx-CeO2对碳烟燃烧的催化作用。因此,MnOx-CeO2复合氧化物具有更高的NO氧化活性和NOx储存能力,更有利于NOx促进碳烟氧化。
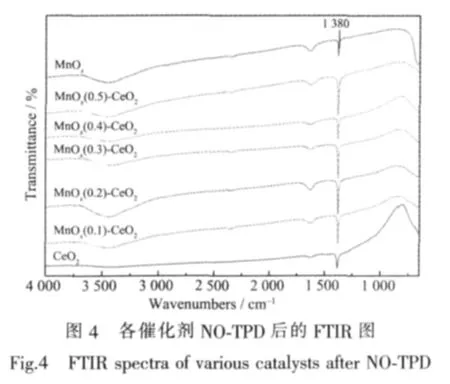
2.4 原位DRIFTs
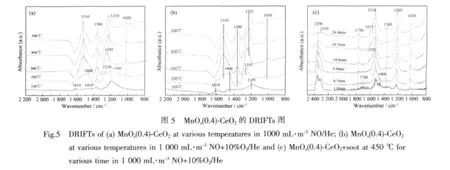
TPO活性测试中发现MnOx(0.4)-CeO2和MnOx(0.5)-CeO2活性相对较高,故利用原位DRIFTs进一步考察MnOx(0.4)-CeO2表面吸附物种,结果如图5所示。
(a)MnOx(0.4)-CeO2在 1 000 mL·m-3NO/He 气氛中的DRIFTs
图5(a)为不同温度下 MnOx(0.4)-CeO2在 1 000 mL·m-3NO/He 中的 DRIFTs图。首先将 MnOx(0.4)-CeO2在 400 ℃、Ar气氛下,进行 0.5h的吹扫,然后降至100℃,采集背景数据,再将预处理过后的催化剂样品置于1 000 mL·m-3NO/He气氛下进行吸附,程序升温速率为10℃·min-1,观察在100~500℃范围内催化剂表面物种的变化。由图5(a)可见,1 380 cm-1处出现明显的吸附峰,归属于相对稳定的离子NO3-[3,17-19,21];吸附峰随着温度的升高而逐渐增大,在500℃达到最大值。1 020 cm-1处的吸附峰于200℃后开始出现,为双齿NO3-[2,17-18,20],其峰强也随温度升高而逐渐增强。其它峰都是在低温下出现至高温下减弱和消失,或是高温下产生新峰。1 195 cm-1处峰归属于金属螯合亚硝酸盐NO2-[3,17-18,20],在低温100℃其峰强最大,但经升温至200℃后减弱,至300℃时消失;1 250 cm-1也是在200℃时出现,300℃时偏移至1 245 cm-1处,为来自于与金属螯合的硝酸盐[2,18]中的硝基-NO2(即NO2+)[2],在400℃时则消失;400℃时1 210 cm-1处出现新峰,即单齿NO3-[17],且到500℃时强度更大。1 410 cm-1为硝基-NO2(即 NO2+)[2-3,17-18],在 200 ℃转化为 1 460 cm-1即单齿 NO2-[18],300 ℃起消失;100 ℃时在 1 610 cm-1处有一吸附峰,归属于气态或弱吸附的NO2[2,17],200℃起消失;300℃起1 510 cm-1出现新峰,归属于单齿NO3-[3,17-19]。由此可见,自100℃起,随着温度的升高,NO2-、NO2+与弱吸附的NO2等经过氧化形成单齿NO3-并储存下来。这揭示了MnOx(0.4)-CeO2具有促进NO的吸附、氧化及NOx储存能力的性质。其中,NO的氧化所用的氧主要来源于催化剂中的氧物种,生成的NO2利于碳烟的氧化[1];当然催化剂表面的氧也可直接参与碳烟的氧化过程[22]。催化剂的氮氧化物储存与还原[23]能力越强,则在反应过程中对碳烟的催化贡献的越多,越有利于促进碳烟燃烧[1]。如 Ito 等[7]发现 MnOx(0.25)-CeO2催化剂在 260℃仍能保持着对NOx的吸附并存储,而NO2作为强氧化剂将提高碳烟氧化速率。
(b)MnOx(0.4)-CeO2在 1 000 mL·m-3NO+10%O2/He气氛中的DRIFTs
为了考察NO在富氧条件下储存在催化剂表面的形态,将 1 000 mL·m-3NO/He 气氛(图 5(a))换成1 000 mL·m-3NO+10%O2/He气氛,其余预处理条件不变,对MnOx(0.4)-CeO2进行催化剂表面NO吸附的DRIFTs测试,结果如图5(b)所示。比较图5(b)和图5(a),发现相同温度下,富氧气氛与无氧气氛的主要特征峰基本类似,富氧气氛对催化剂表面吸附NO 后生成的 NOxδ(x=2,3;δ=-1,0,+1)的各种形态有一定影响,特别是最终强化了NO3-的储存。吸附峰明显且峰强随温度升高而逐渐增强的1 380 cm-1,仍归属于相对稳定的离子NO3-[3,17-19,21]。
至200℃后在1 010 cm-1处开始出峰的为双齿NO3-[2,17-18,20],其峰强也随温度升高而逐渐增强。其它峰都是在温度较低时出现,温度升高后减弱和消失或产生新峰,即预示含氧、含氮物种的转化,与图5(a)情况基本类似,同样是NO2-、NO2+及弱吸附的NO2自100℃起向更稳定的单齿NO3-转化过程,此处不再细述。这揭示了富氧气氛更促进MnOx(0.4)-CeO2对NO的吸附、氧化及NOx储存能力。
因此,可以推断出富氧条件促进NO的氧化继而促进碳烟的氧化可能经历的过程[3,6,9]。首先NO吸附在催化剂表面后,同时气相O2为催化剂中的氧物种(如 O2-,O-与 O2-)[21]提供氧库[5,6,9,18,24]。接着富氧有利于催化剂体相或表面/界面氧物种如超氧O2-、化学弱吸附氧O-甚至是晶格氧O2-的迁移[3,5-6,9,24],用于NO[3,5,19-21]的氧化或NO2-[20-21]的生成与进一步氧化,生成NO3-及NO2-和NO2。其中NO3-数量占优,以单齿NO3-存在,且在高温下仍能保持。另外MnOx(0.4)-CeO2表面还可观察到有较强的离子NO3-吸附峰。总之催化剂对NOx有强的吸附存储性能,且含有较多的氧物种从而促进NO转化为NO3-。
(c)MnOx(0.4)-CeO2+碳烟在 1 000 mL·m-3NO+10%O2/He气氛中的DRIFTs
为了直接观察碳烟在催化剂上反应过程中生成物种的形态及其演变,深入探讨NO促进碳烟催化氧化的作用机理,将 MnOx(0.4)-CeO2按图 5(b)所述条件预处理后,与碳烟混合,置于450℃环境、1 000 mL·m-3NO+10%O2/He 气氛下反应 30 min,连续采集红外光谱,选取不同时刻点的谱图以作比较,观察催化剂表面上物种的生成及形态变化,如图5(c)所示。其结果显示催化剂储存的NOx物种与图5(b)类似;同时出现了一些新峰即碳烟氧化产生的中间物或产物。
图5(c)中新峰1 615 cm-1归属于来自于C-NO2复合物[11]中的羰基[11],1 550 cm-1为 C-NO2复合物[11,25-26]。综合起来,新峰 1615cm-1与 1550cm-1为催化剂上的表面配合物(SCO,surface complex oxide[3]),即C-NO2复合物[11,25-26]。这暗示了NO的氧化及其氧化产物与碳烟的接触及结合,最终可促成碳烟逐渐氧化。1510 cm-1归属于单齿 NO3-[3,17-19],2 310 cm-1及2 356 cm-1则归属于 CO2的伸缩振动[21,27],1.6 min时呈现出较大强度,至19.5 min后逐渐减弱。2 356 cm-1峰先增大后减小,表明碳烟被逐渐氧化。因此,MnOx(0.4)-CeO2在 NO+O2气氛下通过 NO2的强氧化性,对碳烟的氧化活性逐渐体现出来。随着时间的增加即反应的进行,1 380 cm-1处峰即离子NO3-[3,17-19,21]渐弱,同时伴随CO2生成减少,则说明离子NO3-与CO2的生成正相关。1 205 cm-1处峰即与金属螯合的NO3-[2,20]或单齿NO3-[2,17],且随着时间的增加即反应的进行其峰强度越来越大。1 020 cm-1处峰为双齿NO3-[2,17-18,20]或单齿NO3-[3,17,20],其峰强也随温度升高而稍有增强。1 750 cm-1归属于酸酐[3,11],如环酐[25]、内酯[3,11,26]、乙醚[3]、N2O4[3],或离 子 NO3-[21];1 700 cm-1归属于羧基[11]、内酯[26]、羰基[25-26],或双齿CO32-[20]。1750cm-1与 1700 cm-1归属为 C(O)复合物,其最终归宿是CO2[11],因此该表面氧复合物(SOCs)反映了反应路径。1 460 cm-1处的单齿NO2-[18]在反应6.5 min后消失,推测是NO2-结合催化剂表面氧物种(O2-,O-与 O2-)而转化成单齿 NO3-(1 510 cm-1),因为松接触状态下MnOx-CeO2用于碳烟的氧化过程中,其活性氧物种极易由化学吸附氧在Mn-Ce表面上的氧空位产生[24]。
2.5 反应机理
综合图 5及表 1、2和 3,在 NO/He、1 000 mL·m-3NO+10%O2/He气氛中,低温下(100,200 ℃)检测到的硝基NO2+、单齿NO2-、金属螯合亚硝酸盐NO2-(chelating nitrites)、单/双齿 NO3-(1 410 cm-1、1 460 cm-1、1 195 cm-1、1 250/1 245 cm-1) 在温度升高后逐渐转化为离子 NO3-、单齿 NO3-(1 210 cm-1、1 510 cm-1);200~300 ℃后离子 NO3-开始增多,这与图 4 的结果是一致的。
1 000 mL·m-3NO+10%O2/He气氛中加入碳烟后,在450℃下,随着反应的进行,单齿硝酸盐(1 510 cm-1、1 205 cm-1) 峰伴随其它 NOxδ物种的渐弱越来越明显;羧基(1 700 cm-1)在9.8 min后消失;酸酐(1 750 cm-1)逐渐变强;C(O)复合物出现;14.6 min开始出现表面氧复合物(SOCs)如C-NO2复合物(1 550 cm-1、1 615 cm-1),同时 CO2(2 356 cm-1)峰先增大后减小,表明NO转化的同时对碳烟的氧化有促进作用。综上,即 MnOx(0.4)-CeO2在 1 000 mL·m-3NO+10%O2/He气氛下借助NO2的强氧化性促进碳烟的氧化。

主要考虑NO的转化及其促进碳烟氧化的作用,可以推断出1 000 mL·m-3NO+10%O2/He气氛中MnOx-CeO2氧化碳烟的过程,反应路径如图6所示,依次按以下4个阶段进行:
(Ⅰ) 气态O2生成活性氧物种O(O2-、O-与O2-,它们之间可以相互转化[27]),气态的NO则生成活性物种NO*;
(Ⅱ)NO*低温时生成硝酸盐及少量亚硝酸盐,随着反应的进行在高温时分解出NO2*和O-;
(Ⅲ) NO2*继而与碳烟形成C-NO2(ads)复合物[28],再生成碳氧中间物C(O)(ads)与NO*,O2-与碳烟也可形成C(O)(ads),故 C(O)中氧来源于 C-NO2(ads)复合物(间接来自于O2-)或直接来自于O2-,O2-的氧库则是气相中的O2;
(Ⅳ) 因为O-对活性的关键作用[27,29-31],O-与C(O)(ads)最终结合生成CO2。
3 结 论
(1)在引入NO后,用共沉淀法制备的复合氧化物MnOx-CeO2对应碳烟的起燃温度Ti低至250~303℃,远低于无催化剂与单一氧化物CeO2对应的碳烟起燃温度;同样,复合氧化物的Tm(CO2浓度达到最大值时的峰温)也比MnOx的Tm稍低。明显地,MnOx-CeO2的活性优于单一的CeO2和MnOx,NO的存在促进了碳烟的氧化。
(2)MnOx-CeO2表面存在的对NO较强的吸附能力,更易氧化NO和储存NOx。可能的反应机理为,富氧条件下气相O2源源不断地推动MnOx-CeO2中氧物种(如 O2-,O-与 O2-)的形成(含相互转化)与迁移,促成NO或NO2-的氧化,储存的NOx在低温下以硝酸根形式存在,在高温时释放出活性很强的NO2*和O-,继而促进碳烟氧化,其中间产物包括CNO2复合物与C(O)复合物。
[1]Kirill T,Oliver K,Martin E,et al.Appl.Catal.B,2006,64:72-78
[2]Wu X D,Liang Q,Weng D,et al.Catal.Commun.,2007,8(12):2110-2114
[3]Wu X D,Lin F,Xu H B,et al.Appl.Catal.B,2010,96(1-2):101-109
[4]Zhang D J,Murata Y,Kishikawa K,et al.J.Ceram.Soc.Jpn.,2008,116(1350):230-233
[5]Wu X D,Liang Q,Weng D,et al.Catal.Today,2007,126:430-435
[6]Shan W J,Ma N,Yang J L,et al.J.Nat.Gas Chem.,2010,19(1):86-90
[7]Kazuhiro I,Kouji K,Akihisa W,et al.Catal.Commun.,2007,8(12):2176-2180
[8]Fu M L,Yue X H,Ye D Q,et al.Catal.Today,2010,153(3/4):125-132
[9]Machida M,Murata Y,Kishikawa K,et al.Chem.Mater.,2008,20(3):4489-4494
[10]Wu X D,Liu S,Weng D,et al.Catal.Commun.,2011,12(5):345-348
[11]Setiabudi A,Makkee M,Moulijn J A,et al.Appl.Catal.B,2004,50(3):185-194
[12]ZHANG Ming(张 明),FU Ming-Li(付 名 利),WU Jun-Liang(吴军良),et al.J.Chin.Soc.Rare Earth(Zhongguo Xitu Xuebao),2011,29(3):303-309
[13]Debora F,Nunzio R,Guido S,et al.J.Catal.,2003,217(2):367-375
[14]ZHU Yi(朱艺),WANG Jian-Li(王健礼),CHEN Yong-Dong(陈永东),et al.Acta Phys.-Chim.Sin.(Wuli Huaxue Xuebao),2011,27(4):925-931
[15]Debora F,Paolo F,Guido S,et al.Appl.Catal.B,2003,43(3):243-259
[16]He H,Dai H X,Ou Z T.Catal.Today,2004,90(3):245-254
[17]Wu X D,Liu S,Lin F,et al.J.Hazard.Mater.,2010,181(1-3):722-728
[18]Setiabudi A,Chen J,Mul G,et al.Appl.Catal.B,2004,51(1):9-19
[19]Machida M,Masatoshi U,Kurogi D,et al.Chem.Mater.,2000,12:3158-3164
[20]Masato M,Masatoshi U,Kurogi D,et al.J.Mater.Chem.,2001,11:900-904
[21]Li Y J,Lin H,Shangguan W F.et al.Chem.Eng.Technol.,2008,31(1):138-142
[22]LI Qian(李 倩),WANG Zhong-Peng(王 忠 鹏),MENG Ming(孟明),et al.Environ.Chem.(Huanjing Huaxue),2011,30(1):331-336
[23]Robert B,Sotiris E,Alfons B,et al.Appl.Catal.B,2011,101(3-4):682-689
[24]Liang Q,Wu X D,Weng D,et al.Catal.Today,2008,139:113-118
[25]Smith D M,Chungtai A R.Colloids Surf.,A,1995,105(1):47-77
[26]Akhter M S,Chughtai A R,Smith D M.J.Phys.Chem.,1984,88(22):5334-5342
[27]MING Cai-Bing(明彩兵),YE Dai-Qi(叶代启),LIANG Hong(梁红),et al.Acta Scientiae Circumstantiae(Huanjing Kexue Xuebao),2010,30(1):158-164
[28]ZHU Chang-Quan(朱昌权),LIANG Hong(梁红),LI Shu-Hua(李树华),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2011,27(6):1093-1100
[29]FU Ming-Li(付名利),YE Dai-Qi(叶代启),YUE Xiang-Hui(乐向晖),et al.J.Chinese Soc.Rare Earth(Zhongguo Xitu Xuebao),2010,28(2):165-170
[30]Descorme C,Madier Y,Duprez D.J.Catal.2000,196(1):167-173
[31]Liu J,Zhao Z,Xu C M,et al.Appl.Catal.B,2008,78(1):61-72
Soot Oxidation in NO Atmosphere via MnOx-CeO2Prepared with Co-precipitation
FU Ming-Li1,2WANG Ke-Liang1YU Run-Peng1WEN Xian-Er1ZHANG Ming1HUANG Bi-Chun1WU Jun-Liang1YE Dai-Qi*,1,3LIANG Hong4
(1College of Environmental Science and Engineering,South China University of Technology,Guangzhou 510006,China)
(2Key Lab of The Education Ministry of China for Pollution Control and Ecosystem Restoration in Industry Clusters,South China University of Technology,Guangzhou 510006,China)
(3Guangdong High Education Engineering Technology Research Center for Air Pollution Control,Guangzhou 510006,China)
(4College of Chemistry and Chemical Engineering,Guangzhou University,Guangzhou 510006,China)
MnOx-CeO2with various Mn/Ce ratios were prepared by co-precipitation method,and the results of temperature programmed oxidation (TPO)test revealed that,when exposed to 1 000 mL·m-3NO and 10%O2,ignition temperature Tifor soot oxidation in presence of MnOx-CeO2ranged in 250~303 ℃,much lower than the Ti(402℃)without catalyst,the Ti(334℃)in presence of CeO2and the Ti(346~360℃)in presence of MnOx-CeO2without NO,which also corresponded ~to the case of MnOxapproximately.However,the combustion temperature at maximum rate Tm(413~441 ℃)of MnOx-CeO2was lower than that of MnOx(441 ℃).Apparently,NO promoted the oxidation of soot,and MnOx-CeO2exhibited higher activity than CeO2and MnOxalone.NOTPD,FTIR and in situ DRIFTs showed that MnOx-CeO2was greatly adsorptive to NO,enhancing NO oxidationand NOxstorage capacity,as well as the oxidation of soot.The possible mechanism could be that gaseous O2in rich O2condition accelerated the formation(including transformation)and mobility of oxygen species in catalyst(such as super oxygen O2-,weakly chemisorbed oxygen O-,and lattice oxygen O2-),facilitating the oxidation of NO and NO2-;The NOxwas stored in the form of NO3-at low temperatures,and NO3-easily decomposed into highactivity NO2*and O-at higher temperatures,thus soot oxidation was promoted.The intermediate products included composite C-NO2and composite C(O)complex.
NO;MnOx-CeO2;soot oxidation;catalyst;co-precipitation
O614.33+2;O643.36+1;X506;X513
A
1001-4861(2012)08-1593-08
2012-01-01。收修改稿日期:2012-03-12。
国家自然科学基金(No.51108187,No.50978103);广东高校科技成果转化项目 (No.cgzhzd0803);中央高校基本科研业务费(No.2012ZM0041,No.2011ZM0048)资助项目。
*通讯联系人。E-mail:cedqye@scut.edu.cn
