基于多基因组合选择模型的结肠癌特征基因选取
2012-09-11马超
马 超
【统计应用研究】
基于多基因组合选择模型的结肠癌特征基因选取
马 超
(南京大学公共卫生管理与医疗保障政策研究中心,江苏南京210093)
通过基因的Bhattacharyya距离指标过滤掉大部分无关基因,然后探索性的提出了一种建立多基因组合选择模型的统计方法。从候选特征基因中选取了8个可能的结肠癌特征基因集合,判别分析的结果证明了该方法的可行性。
基因表达谱;生物信息学;多基因组合选择模型;结肠癌
一、引 言
DNA微阵列(DNA microarray),也叫基因芯片,是最近数年发展起来的一种能快速高效检测基因表达水平、DNA片段序列的新技术。这样生物学家通过并行检测成千上万个基因的表达数据,在基因组水平上比较正常和疾病下基因表达的差异,找出决定样本类别的一组基因“标签”,即“信息基因”(informative genes),这是正确识别肿瘤类型、给出可靠诊断以及简化实验分析的关键所在,同时也为抗癌药物的研制提供了捷径。对肿瘤基因表达谱进行有效分析,挖掘和发现其中蕴含的知识,是当前生物信息学研究的重点课题[1-5]。
通常基因数目很大,在判断肿瘤基因标签的过程中,需要剔除掉大量“无关基因”,从而大大缩小需要搜索的致癌基因范围。Golub等以“信噪比”(Signal to noise ratio)作为衡量基因对样本分类贡献大小的指标,运用加权投票的方法对亚型进行识别,仅根据72个样本,就从7 129个基因中选出了50个可能与亚型分类相关的信息基因,富有创造性[1]。Guyon等人则利用支持向量机的方法,再从中选出了8个可能的信息基因[6]。Alon等人利用层次聚类法对结肠癌样本数据进行了分析研究,选出含有2 000个特征基因的数据集合[7]。在Alon的研究基础上,Zhang等人通过递归分割树的方法,归纳出2个特征基因集合[8];李霞等人使用集成决策的方法,得到3个特征基因集合[9]。李颖新等人则采用了模糊识别方法进行分析[10];刘全金等人采用了浮动顺序搜索算法,并以RBF支持向量机作为分类器[11];何爱香采用了遗传算法和CFS算法选择基因子集[12]。
上述方法均具有一定的参考价值,且鉴别出的基因也具有较高的准确率,但是方法相对复杂。本文基于前人的研究,在支持向量机的方法上做了一些改进,创新性的提出了多基因组合选择模型,提取出了相关信息基因,并且该方法的正确率高达95%。
二、数据来源与预处理
本文的实验数据来自Alon公布的结肠癌基因表达谱数据集,包含40个结肠癌组织样本和22个正常组织样本,每个样本包含2 000个基因的表达数据。该数据集维数为2 000,远大于样本数62。因此,首先有必要对数据进行过滤,剔除大量无关基因,缩小需要搜索的致癌基因范围。
本文采用Bhattacharyya距离来衡量基因含有样本分类信息的多少。以两样本为例,Bhattacharyya距离体现了属性在两个不同样本中分布的差异。具体形式见式(1):

其中B是基因的Bhattacharyya距离。由式(1)可知,Bhattacharyya距离由两部分构成:第一项表示基因在两个类别中分布均值的差异对样本分类的贡献;第二项表示两个类别中分布方差的不同对分类的贡献。依据该距离公式,只要分布的方差出现大的差异,仍然可以获得较大的距离值。从模式分类的角度看,基因的Bhattacharyya距离越大,利用该基因的信息,样本的可分性就越好。根据式(1),计算了每个基因的Bhattacharyya距离,并做出了基因的Bhattacharyya距离分布的直方图,见图1。
依据基因所含样本类别信息的多少,将基因分为“信息基因”和“无关基因”两类。设SI为信息基因集合,Sn为无关基因集合,则“信息基因”与“无关基因”可定义如式(2):

图1 基因的Bhattacharyya距离分布的直方图

其中g为基因,B(g)为基因g的Bhattacharyya距离,q为指定的Bhattacharyya距离的阈值。
由图1可知,94%的基因的Bhattacharyya距离小于0.1。故取阈值为0.1。这些基因在两个类别中的分布,无论其均值还是方差均无明显差异,可以作为无关基因剔除。
肿瘤基因表达谱中基因Bhattacharyya距离的详细分布情况见表1。依据表1和式(2)对信息基因和无关基因的定义知:取阈值q=0.1,card(SI)=115,即在2 000个基因中,有115个基因为信息基因;card(Sn)=1 885,即有1 885个基因为无关基因。SI中115个基因均在不同程度上包含了样本的分类信息,是进一步分析的基础。

表1 基因的Bhattacharyya距离分布情况表
三、多基因组合选择模型
癌症的发病不仅跟单个基因有关,更多情况下与多条基因的综合作用有关。因此,我们将在上文剔除无关基因的基础上建立多基因组合选择模型,以识别其中的信息基因,即确定基因标签。具体步骤如下:
(一)分类因素指标的确定
仍采用Bhattacharyya距离公式,当基因个数大于等于2时,多维Bhattacharyya距离公式如下:

其中J(Fi)表示含有i个基因的特征子集Fi的Bhattacharyya距离。μ1、μ2为特征子集Fi中的基因在正常人和癌症患者中分布的均值向量,∑1和∑2为相应的协方差矩阵。
(二)两个基本基因的查找
利用MATLAB程序求出这115条基因中的maxJ(F2),并确定相应的基因号,程序如下:

经过C2115次计算,得到结果为:maxJ(F2)=1.443 4,相应的基因集合为F2-max={1 325,1 967}。
(三)最优特征子集中的基因个数k*的确定
定义:y(k)=maxJ(Fk),则y(k)为一递增函数,令Δy(k)=y(k+1)-y(k),理论上,当Δy(k*)→0时,则k*为所确定的最优基因个数。
执行过程及算法如下:
先固定步骤2中两个基本的基因,再顺序搜索逐一添加剩余基因数至k*,算法为:
1.SI=SI-Fi-max(i=2,…,115)
2.搜索g∈SI,使F(i+1)-max={Fi-max,g}的评价值J(F(i+1)-max)最大。
经过(113+112+…+98)=1 688次计算,结果见表2:
从表2可以看到多维Bhattacharyya距离的最大值maxJ(Fi)关于基因数i成递增趋势。
但是,如图2所示,多维巴氏距离最大值的递增趋势随着i的增加有所减缓。考虑到癌症的发病往往是跟少数基因有关,因此k*一方面不会很大,另一方面由图2的特征,我们取最优特征子集中的基因个数k*=18。

图2 多维巴氏距离最大值图

表2 Fi-maxJ1(Fi)表
(四)基因标签的初步确定
为了初步确定基因标签,我们先设定标签中的基因数,然后从表2所示的全部18个基因中寻找不同基因数所对应的基因标签。程序如下:
function f=ppp(x,y);
y=[1 325,1 967,1 750,1 671,1 381,698,
1 843,1 473,1 549,822,1 892,1 411,
1 668,245,1 221,1 770,1 511,1 346];
k=nchoosek(y,18);
for i=1:size(k,1);
a=x(k(i,:),:);
u1=mean(a(:,2:23)');%u1为行向量
u2=mean(a(:,24:end)');
v1=cov(a(:,2:23)');
v2=cov(a(:,24:end)');
bb=0.125*(u2-u1)*inv(v1/2+v2/2)*(u2-u1)'+0.5*log((det(v1/2+v2/2))/sqrt(det(v1)/det(v2)));
b(i,:)=[k(i,:),bb];
end
f=b;
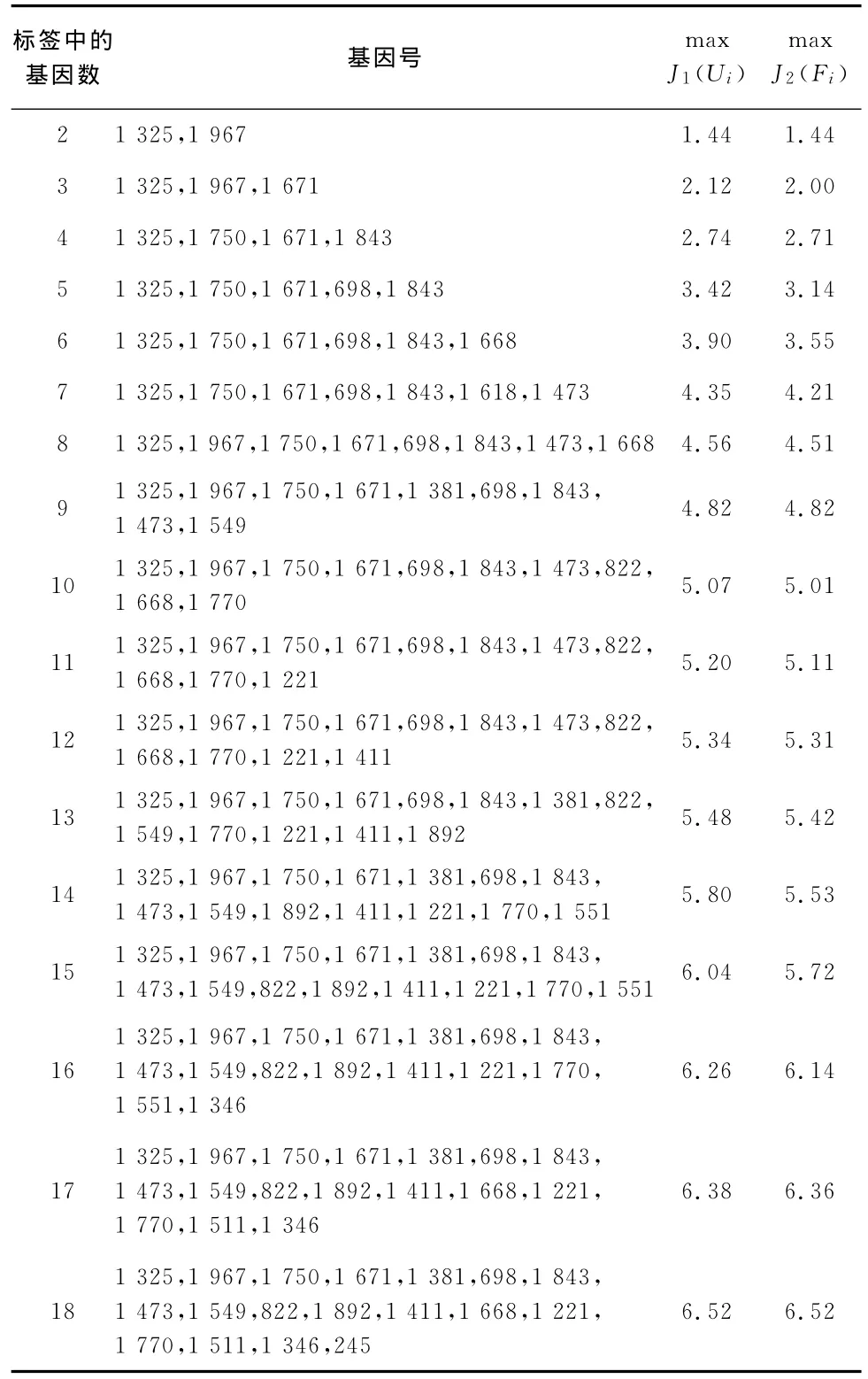
表3 Ui-maxJ2(Ui)表
从表3的后两列数据可知,由于循环时每一步均在18个基因中全局查找,因此

根据表3的结果,统计每个基因所出现的频数,见表4:

表4 信息基因频数表
根据频数表,我们初步认定基因标签中应含有如下6条基因,见表5:

表5 基因标签表
(五)基因标签的最终确定
以上述6条基因为基础,然后从表4剩余的基因中逐一添加,并从SI的那115条基因中任意挑选s条基因加入。求解最大Bhattacharyya距离值maxJ3(Hi),经过多次试算,并结合表1中Δy(k)的值及图2,发现S的合理取值为2。程序如下:
function f=ppp6(x,y);%固定6条
a=setdiff(y(:,1),[1325 1671 1750 1843 698 1967])';
k(:,1:2)=nchoosek(a,2);
k(:,3)=1325;
k(:,4)=1671;
k(:,5)=1750;
k(:,6)=1843;
k(:,7)=698;
k(:,8)=1967;
for i=1:size(k,1);
a=x(k(i,:),:);
u1=mean(a(:,2:23)');%u1为行向量
u2=mean(a(:,24:end)');
v1=cov(a(:,2:23)');
v2=cov(a(:,24:end)');
bb=0.125*(u2-u1)*inv(v1/2+v2/2)*(u2-u1)'+0.5*log((det(v1/2+v2/2))/sqrt(det(v1)/det(v2)));
b(i,:)=[k(i,:),bb];
end
f=b;
经过C2109次计算,结果表明,当基因标签中的基因个数为8时,max J3(Hi)=max J2(Ui)=4.562 6。类似上面的做法,继续从表4中选取累计频数最大的7条基因,再从全局搜索2个基因,搜到的9个基因与表3中的显示完全一样,经过验证,当i≥8时,max J3(Hi)=max J2(Ui),且相应的特征基因子集也完全相同,由此证明,表5的选取是合理的,所以最终的基因标签见表6。
在此结果上,本研究通过fisher判别分析对结果进行评价,将62个样本中42个分为训练集,另外20个作为测试集,每个样本包含上述8个基因的基因表达水平。测试集中20个样本仅有1个错判了,正确率高达95%。总体来看上述多基因组合选择模型的效果很不错。

表6 最终基因标签表
四、总 结
本文在前人研究的基础上,对支持向量机的方法做了一些改进,从统计学角度创新性的提出了一种提取信息基因的新方法——多基因组合选择模型,并鉴别出了8个信息基因,该方法的正确率高达95%。与传统方法相比,不仅提高了诊断的正确性,而且降低了诊断方法的复杂性。这种新的方法经过逐步调试及改进,将会成为很多传统方法的一个重要补充,并能起到验证的作用,对于癌症的临床诊断以及生物医学研究提供一定的参考作用。
[1] Golub R R,Slonim D K,Tamayo P.Molecular Classification of Cancer:Class Discovery and Class Prediction by Gene Expression Monitoring[J].Science,1999,289(15).
[2] Khan J,Wei J S,Ringner M.Classification and Diagnostic Prediction of Cancers Using Gene Expression Profiling and Artificial Neural Net Works[J].Nature Medicine,2001,7(6).
[3] Furey T S,Cristianini N,Duffy N,et al.Support Vector Machine Classification and Validation of Cancer Tissue Samples Using Microarray Expression Data[J].Bioinformatics,2000,16(10).
[4] Ramaswamy S,Golub T R.DNA Microarrays in Clinical Oncology[J].Journal of Clinical Oncology,2002,20(7).
[5] Wang Y,Makedon F,Ford J C.Hykgene:A Hybrid Approach for Selecting Marker Genes for Phenotype Classification Using Microarray Gene Expression Data[J].Bioinformatics,2005,21(8).
[6] Guyon I,Weston J,Barnhill S,et al.Gene Selection for Cancer Classification Using Support Vector Machines[J].Machine Learning,2000,46(13).
[7] Alon U,Barkai N,Notterman D A.Broad Patterns of Gene Expression Revealed by Clustering Analysis of Tumor and Normal Colon Tissues Probed by Oligonucleotide Arrays[J].Proc Natl Acad Sci Usa,1999,96(7).
[8] Zhang H,Yu C Y,Singer B.Recursive Partioning for Tumor Classification with Gene Expression Microarray Data[J].Proc Natl Acad Sci Usa,2001,98(12).
[9] 李霞,饶绍奇,张田文.应用DNA芯片数据挖掘复杂疾病相关基因的集成决策方法[J].中国科学C辑:生命科学,2004,34(2).
[10]李颖新,刘全金,阮晓钢.急性白血病的基因表达谱分析与亚型分类特征的鉴别[J].中国生物医学工程学报,2005(24).
[11]刘全金,李颖新,阮晓钢.基于基因表达谱的结肠癌特征基因选取[J].昆明理工大学学报,2006,36(1).
[12]何爱香.基于遗传算法的结肠癌基因选择与样本分类[J].计算机工程与应用,2007,48(18).
Informative Genes Selection of Colon Cancer Based on Polygenic Combination Selection Model
MA Chao
(Center for Health Management and Care Security Policy Research,Nanjing University,Nanjing 210093,China)
To select informative genes of colon cancer by analysis of gene expression.Most irrelevant gene filtration by the gene distance method of Bhattacharyya,then putting forward a statistical method of establishing polygenic combination selection model.The study selects 8possible informative genes sets of colon cancer from the candidate informative genes.The results of discriminant analysis show the feasibility of this approach,and play a certain reference arriving role to clinical diagnosis of cancer and research in biomedical sciences.
DNA microarray;Bioinformatics;polygenic combination selection model;colon cancer
book=78,ebook=27
Q16∶TP181
A
1007-3116(2012)06-0078-05
(责任编辑:马 慧)
2011-11-25
马 超,男,江苏省南京人,博士生,研究方向:卫生经济与医疗管理。
