基于变长子串的DNA重复序列预归并屏蔽方法
2012-09-08杨进才
蔡 葵,杨进才
(1.武汉理工大学华夏学院,湖北武汉 430223;2.华中师范大学计算机科学系,湖北武汉 430079)
针对 DNA 序列拼接,REPS[1]方法通过度量定长子串来确定重复序列。文献[2-3]以此为基础,提出了各自的定长为k的重复序列识别屏蔽方法。文献[4]进一步提出了预归并的思想,但仍然局限于定长子串识别方法,对偏长的DNA重复序列处理能力不强,识别不够精确。
笔者在PreMerged方法的基础上,引入HUANG[5]等提出的 Superword array 概念,继续预归并思想,提出了利用变长子串来识别且屏蔽重复序列的VPreM方法。模拟实验证明,较之Pre-Merged方法,VPreM方法识别重复序列的精确度更高,并且可以更大规模地缩减shotgun集合规模。
1 基于变长子串的预归并算法
1.1 基本思想
将shotgun集合中所有fragments片段末尾加上字符#,并串连成一大字符串F,为F中每个字符按顺序编号。由字符集{A,G,C,T}上w个字符组成的字符串,就是一个长为w的word,长为w的word一共有4w个。将这4w个word按字母顺序编号,形成一个word字查找表。表1为w值为2的word字查找表。

表1 w为2的word字查找表
大字符串 F上以p位置起始,长为 w的word,其在查找表中的编号Nu,就是p位置的编码,记为Code(p)=Nu。若word中含#,则编号统一设为Nu=-1。图1为一个大字符串F编码示意图。
通过编码可以找出任意两个fragments之间,连续h个长为w的word组成的重复部分repeats。假设shotgun集合中fragments总数目为n,可以推导出h及w的取值满足h×w≥log4n。确定h和w的值之后,就可以为大字符串F编码。然后根据不同fragments之间编码的连续相同性(变长子串),来进行重复序列识别及预归并屏蔽工作。

图1 大字符串F编码示意图
1.2 构建h×w变长子串统计表S
h×w变长子串统计表 S={Pos,FragNum,Loc,CodeArray},其中Pos为字符在F中的位置;FragNum为该字符所属fragment的编号;Loc为该字符在所属fragment中的位置;CodeArray={Code(Pos),Code(Pos+w),…,Code[Pos+(h-1) ×w]},存储连续 h个长为 w的 word编码。按CodeArray值排序后,对应图1的h×w变长子串统计表S1如表2所示。
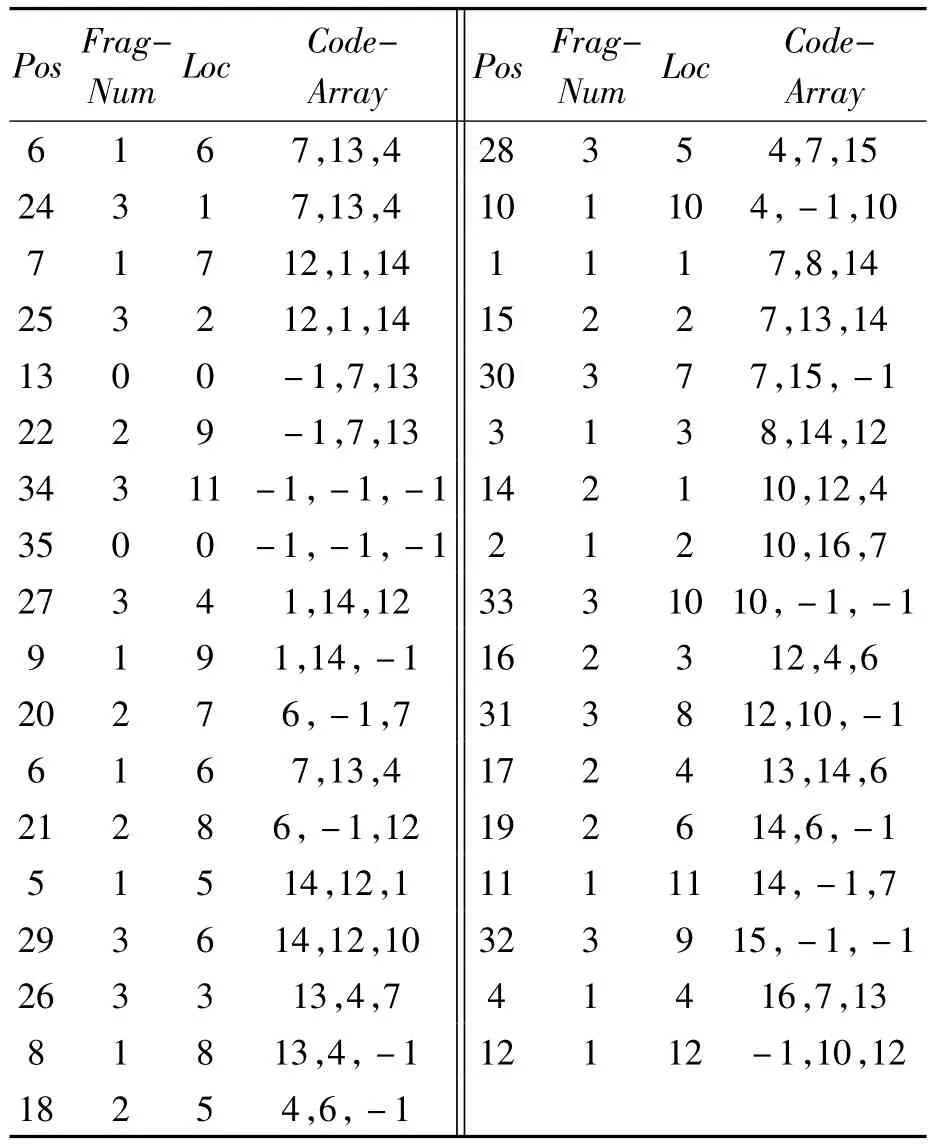
表2 h×w变长子串统计表S1
先在表S1中去除所有CodeArray值中含-1的记录,再去除仅部分与CodeArray值相同的记录,增加FR项表示相同CodeArray值个数。得到优化后的h×w变长子串统计表S2,如表3所示。
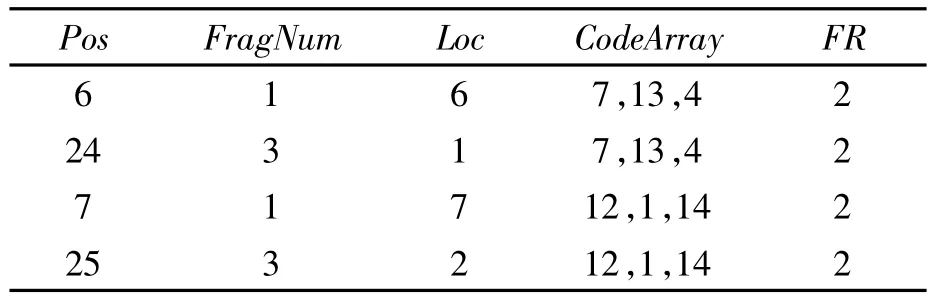
表3 优化后的h×w变长子串统计表S2
设shotgun集合是DNA目标序列的C条克隆随机打散而成,其阈值T=C。可认为表S2中FR值不超过T对应的子串不属于某个repeats;反之则认为属于某个repeats。
1.3 变长子串的直接预归并
从表S2中取出FR值没有超过阈值T且CodeArray项值相同的记录,根据记录中的Frag-Num和Loc值,直接在 shotgun集合 FRAG[n]中预归并这些fragments为一个新的更大的fragment。其算法如下:

1.4 预归并及识别重复序列
在表S2中去除所有FR值处于阈值T之内的记录,则剩余记录均为与repeats相关的变长子串信息。根据每条记录中的FragNum和Loc值,可以分别在shotgun集合中向左、向右预归并对应fragments,并借此识别出包含同一h×w变长子串的repeats的边界。其算法如下:
i=1;
while(表S2未结束){
取表S2的第i条记录,记为Si;
取Si的FR值,记为Time;
//以下为往左分析repeats信息
取第i至第i+Time-1条记录中Loc值最大的记录,记为St;
将St对应的fragment编号记入NL数组;
取St的Pos值,记为Pt;
For(表S2的第i+1条至第i+Time-1条记录){
取当前记录Sc的Pos值,记为Pc;
以 Code[Pc]与 Code[Pt]为起点,同步往左逐个比较Code值;
遇到第一个非负且不同的Code值,将Sc对应的fragment编号记入NL数组;
截取Si与 Sc对应 fragment从起点到不同Code值位置片段,放入RepL集中;
截取Sc对应fragment从不同Code值位置开始到Pc+h×w-1位置片段,记为REL;
}
//以下为往右分析repeats信息
取第i至第i+Time-1条记录中对应fragment长度减Loc值最大的记录,记为St;
将St对应的fragment编号记入NR数组;
取St的Pos值,记为Pt;
For(表S2的第i+1条至第i+Time-1条记录){
取当前记录Sc的Pos值,记为Pc;
以 Code[Pc+h×w]与 Code[Pt+h×w]为起点,同步往右逐个比较Code值;
遇到第一个非负且不同的Code值,将Sc对应的fragment编号记入NR数组;
截取Si与Sc对应fragment从不同Code值位置到终点片段,放入RepR集中;
截取Sc对应fragment从Pc+h×w开始到不同Code值位置片段,记为RER;
}
识别出第i至第i+Time-1条记录中的Repeats=REL+RER;
顺序提取 NL中的 fragment编号 u,更新FRAG[u]=RepL中对应的左边界;
顺序提取 NR中的 fragment编号 v,更新FRAG[v]=RepR中对应的右边界;
i=i+Time;
}
1.5 方法的计算复杂性分析
h×w变长子串统计表S是通过扫描大字符串F得到的,其复杂性为Ο(l·n·h·4w),其中l为片段平均长度,n为shotgun集合中片段总数目,h为连续word的个数,w为字word的长度。原始表S经优化后,其中记录数降为[T·4(h·w)](T为阈值),随后的一系列预归并及shotgun片段更新的复杂性,均在Ο(l·n·T)之内,因此综合得到的预归并复杂性为Ο(l·n·T2·4(h·w))。
将其与文献[2-3]方法的复杂性Ο(l·n·4k)相比较,由于h×w的值可视为与k相当,因此笔者提出的方法复杂性比文献[2-3]中的方法多了T2项,略大。再与文献[4]方法的复杂性Ο(l·n·42k)比较,由于T2比4k小,因此笔者提出的方法复杂性比文献[4]方法稍小。实质上,l、T、h和 w均是有定值的,以上方法的复杂性最终都可以转化为Ο(n)。笔者提出的方法是通过变长子串来识别repeats,识别精确性有所提高,对fragments的预归并能力较之定长子串方法也有所增强。
2 计算机模拟分析
笔者提出的算法命名为VPreM,使用VC语言编程,DNA目标序列和shotgun集合均用SQL Server 2003存储,所有模拟实现均在Intel Core2 E4300(1.8 GHz),1 G内存(667 MHz)的PC机上完成。选取与文献[4]相同的5条DNA序列来做模拟分析。根据克隆条数C和分解片段数目n,确定各个关键值:T=C;k=[log4n]+2;w=3;h=[k/w]+1。
用VPreM、PreMerged以及当前流行的RECON[6]方法分别处理以上5个 shotgun集合,最终识别结果如表4所示。3种方法的各自运行时间如图2所示。
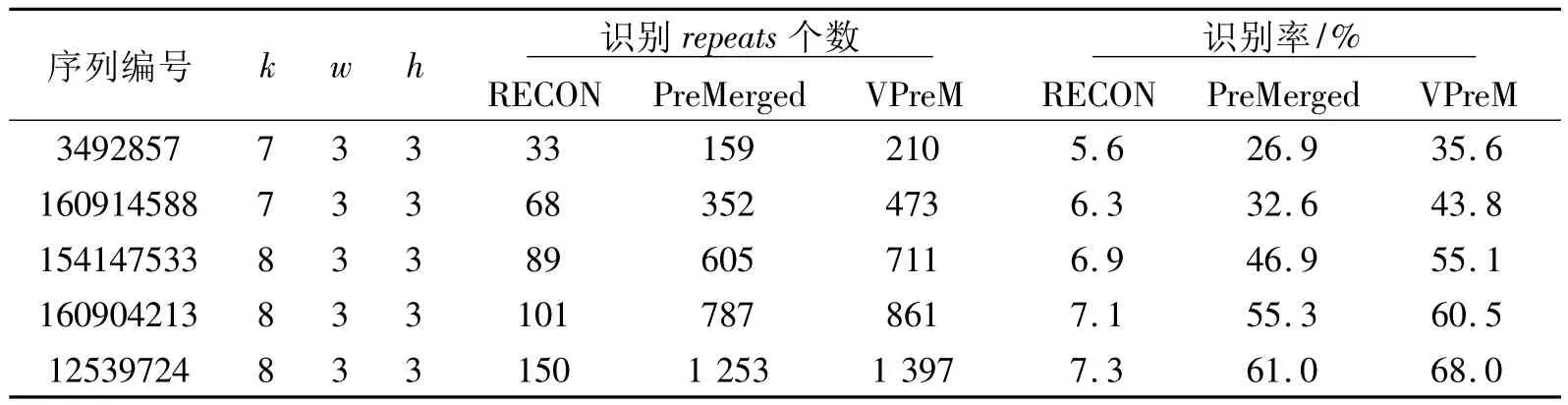
表4 VPreM、PreMerged和RECON的识别结果
从表4可以看出,笔者提出的VPreM方法,在识别重复段的能力上明显强于RECON,比Pre-Merged略强。由图2可知VPreM方法的计算速度稍稍优于PreMerged,更优于RECON。
将以上5个shotgun集合分别用VPreM、Pre-Merged、RECON 3种方法预处理后,再交给Phrap拼接,运行所耗费时间比较如图3所示。

图2 VPreM、PreMerged和RECON的运行时间

图3 分别用VPreM、PreMerged和RECON预处理后Phrap拼接时间比较
由图3可以观察到,经过VPreM方法预归并再用Phrap工具进行拼接,较之采用PreMerged和RECON两种方法预处理后的shotgun集合,具有更快的速度,这也进一步验证了笔者所提出方法的正确性。
3 结论
VPreM方法充分利用h×w的灵活可变性,能精确识别出不同长度repeats的边界,较之基于定长子串的识别方法,效率更高;与预归并方法的结合,也使得识别过程中获取的信息得到了更充分利用。可以应用于文献[7-10]等对应的拼接算法的前期处理工作之中,对提高拼接效率,减少拼接错误,有一定的积极意义。
[1]WANG J.REPS:a sequence assembler that masks exact repeats identified from the shotgun data[J].Genome Research,2002(12):824-831.
[2]张博峰,王正华.DNA片段拼接中基于定长特征子串的重复序列信息屏蔽方法[J].国防科技大学学报,2002,24(6):67-70.
[3]王磊,张祖平,陈建二.DNA片段拼接中重复序列算法研究[J].计算机科学,2006,33(7):164-170.
[4]蔡葵,杨进才.DNA片段拼接中的预归并重复序列屏蔽方法[J].计算机工程,2009,35(4):88-90.
[5]HUANG X Q,YANG S P.Application of a superword array in genome assembly[J].Nucleic Acids Research,2006,34(1):201-205.
[6]BAO Z R.RECON Documentation[EB/OL].[2011-07-08].http://selab.janelia.org/recon.html.
[7]PHIL G.Phrap documentation[EB/OL].[2011-07-08].http://www.phrap.org/phredphrapconsed.html.
[8]GRANGER G S,OWEN W.TIGR assembler:a new tool for assembling large shotgun sequencing projects[J].Genome Science & Technology,1995,1(1):9-19.
[9]HUANG X Q,ANUP M.CAP3:A DNA sequence assembly program[J].Genome Research,1999(9):868-877.
[10]PEVZNER P A,TANG H X,WATERMAN M S.An eulerian path approach to DNA fragment assembly[J].Proceedings of National Academy of Sciences,2001,98(17):9487-9753.
