左旋西替利嗪二盐酸盐合成工艺的优化
2012-07-28徐小军尤庆亮赵蒙蒙喻宗沅
徐小军,尤庆亮,赵蒙蒙,喻宗沅
(湖北省化学研究院,湖北 武汉 430074)
左旋西替利嗪二盐酸盐(Ⅵ)是一种高效、安全的抗过敏药物,也是一种高选择性外周H1受体拮抗剂,2001年2月首次在德国上市,在三大抗过敏药物中最安全、适用范围最广,作用时间长达24 h,起效剂量低,是FDA认可的可以安全用于孕妇的抗过敏药物。与西替利嗪二盐酸盐相比,具有无嗜睡、无镇静作用等突出优点。
Corey等[1]以左旋4-氯二苯甲胺为原料,用N,N-二氯乙基-4-甲基苯磺酰胺构建哌嗪环,得到左旋1-N-单对氯二苯甲基-4-N-对甲苯磺酰基哌嗪,再与HBr/冰醋酸和苯酚反应,除去对甲苯磺酰保护基,得到高光学纯度的左旋4-氯二苯甲基哌嗪,收率为59%,该方法需用大量的溶剂萃取以除去粗品中的苯酚衍生物。王玉玲等[2]优化了反应条件,以对羟基苯甲酸代替苯酚,到反应终点时,直接将反应混合物倾入水中,经过滤除去对羟基苯甲酸的衍生物,可得到高光学纯度的左旋4-氯二苯甲基哌嗪,收率为62%;左旋4-氯二苯甲基哌嗪与2-(2′-氯乙氧基)乙酰胺反应得到相应的酰胺,再经水解得到目标化合物左旋西替利嗪。Rajan等[3]用N,N-二氯乙基-4-甲氧基苯磺酰胺代替N,N-二氯乙基-4-甲基苯磺酰胺,不需添加对羟基苯甲酸,形成哌嗪环后去保护基条件比较温和,简化了操作程序,左旋4-氯二苯甲基哌嗪纯度可达99%,产率84.2%。
作者参照文献[2~5],以L-(+)-酒石酸为拆分剂对4-氯二苯甲胺(Ⅰ)进行拆分,制得左旋4-氯二苯甲胺(Ⅱ);再参照文献[3]的方法以N,N-二氯乙基-4-甲氧基苯磺酰胺与化合物Ⅱ反应,得到左旋4-氯二苯甲基哌嗪(Ⅳ);左旋4-氯二苯甲基哌嗪乙醇(Ⅴ)、左旋西替利嗪二盐酸盐(Ⅵ)的缩合和成盐反应参照文献[2~7]进行,其合成路线见图1。
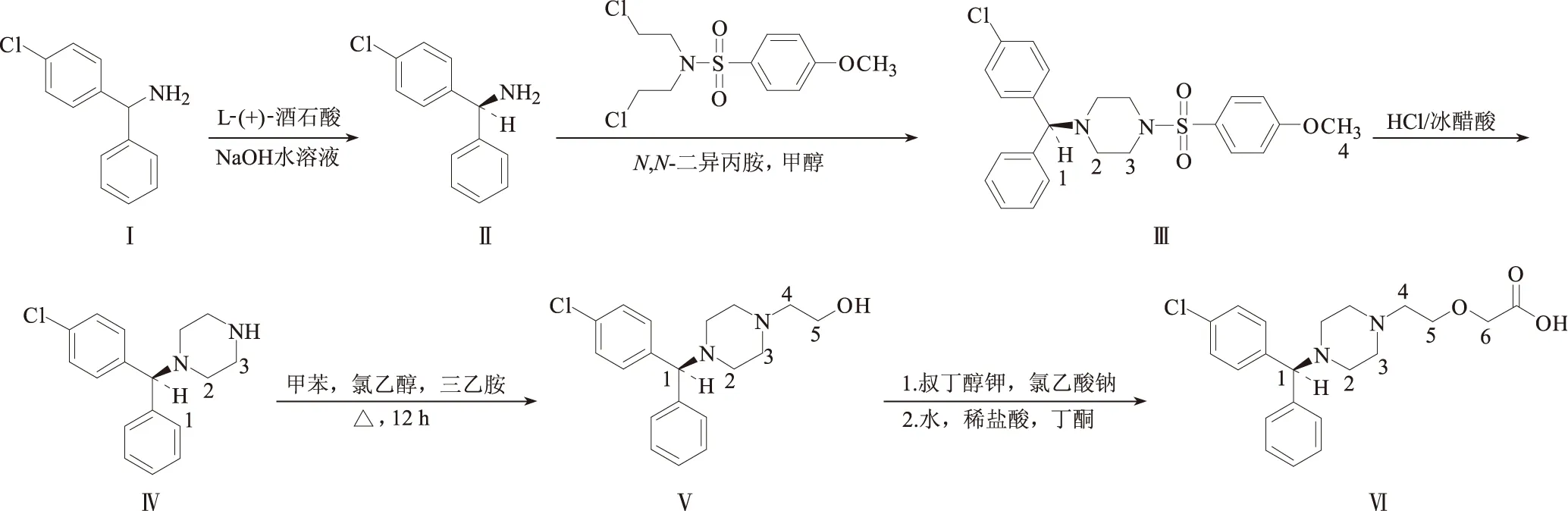
图1 左旋西替利嗪二盐酸盐的合成路线
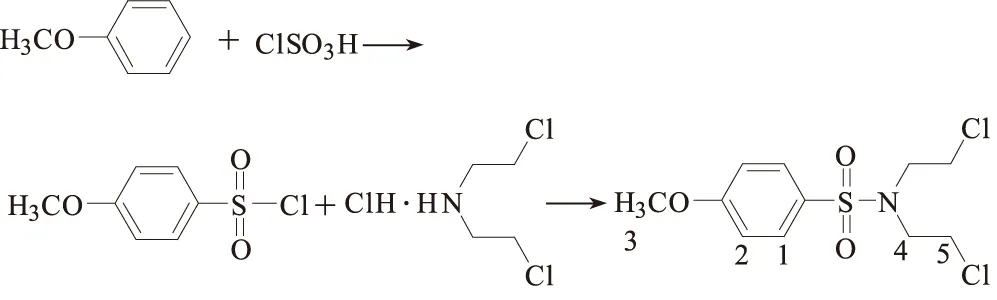
其中,N,N-二氯乙基-4-甲氧基苯磺酰胺的合成路线如下:
1 实验
1.1 试剂与仪器
集热式磁力搅拌器;SHZ-D(Ⅲ)型循环水式真空泵;DZ-1BC型真空干燥箱;电子天平(精确度0.01 g);PTHW型电热套;RE52CS型旋转蒸发仪;Prostar230型高效液相色谱仪,美国Varian公司;254 nm紫外分光仪;核磁共振波谱仪,德国Bruker公司;SGWX-4型显微熔点仪;NEXUS 470型傅立叶红外分光光度仪,美国Nicolet公司;旋光仪,上海亚荣生化仪器厂。
1.2 方法
1.2.1 目标化合物的合成
(1)左旋4-氯二苯甲胺(Ⅱ)的合成
取6.4 g(0.043 mol)L-(+)-酒石酸溶于30 mL水中,搅拌下加入9.0 g(0.041 mol)混旋4-氯二苯甲胺盐酸盐(NaOH中和去除盐酸),搅拌加热至全溶后冷至常温,有白色固体析出,抽滤,水洗;在滤饼中加入90 mL水、2.5 g活性炭,加热搅拌回流1 h,冷至80 ℃,过滤,滤液冷却至室温,抽滤;滤饼再用水重结晶,抽滤,转入烧瓶,加30 mL水搅拌成浆状,冷却下滴加5 mL 30% NaOH水溶液,继续搅拌约2 h;转入分液漏斗中;用30 mL石油醚萃取3次,油层用饱和食盐水洗涤后加无水Na2SO4干燥过夜,滤去Na2SO4,蒸出石油醚,即得化合物Ⅱ。
(2)左旋[4-(-氯苯基)苯甲基]-4-[(4-苯甲氧基)磺酰]哌嗪(Ⅲ)的合成
取30.0 gN,N-二异丙胺、13.0 gN,N-二氯乙基-4-甲氧基苯磺酰胺、8.1 g化合物Ⅱ置于500 mL烧瓶中,升温至127 ℃,回流10 h,反应完毕,降温至80~90 ℃,加入30 mL甲醇,再降温至0~5 ℃,继续搅拌30~60 min,抽滤,滤饼用10 mL甲醇洗涤,将甲醇蒸去一半后冷却结晶,50~60 ℃真空干燥,即得化合物Ⅲ。
随着高校教育事业的迅速发展,高校毕业生每年都在不断增长,高校培养出的优秀毕业生正在走向社会,越来越多的高校教师顺应市场的需求也出现人才流动,教师在工作中取得的科研成果也正在被转化成产能应用到社会的各个领域。高校档案馆在这场变革中应该积极发挥档案馆的作用,不能停留在传统的服务功能上,而要提升创新服务的能力,不断满足师生对高校档案资源的新需求,实现高校档案馆更高的服务价值。
(3)左旋4-氯二苯甲基哌嗪(Ⅳ)的合成
将10.0 g化合物Ⅲ缓慢地加到饱和HCl的冰醋酸中,加热至70~75 ℃,反应3 h后,冷至25~35 ℃,加水52 g,搅拌15~20 min,反应混合物用甲苯(2×30 mL)洗涤,将水层冷却至0~5 ℃,用40%的NaOH溶液调水层pH值至11~12,用甲苯(2×30 mL)萃取有机层,用纯净水洗涤,浓缩,浓缩物加入50 mL水,于25~35 ℃下搅拌2~4 h,抽滤,滤饼再用少量水洗涤,于30~35 ℃下真空干燥,即得化合物Ⅳ。
(4)左旋4-氯二苯甲基哌嗪乙醇(Ⅴ)的合成
取14.3 g(0.050 mol)化合物Ⅳ、70 mL干燥的甲苯、6 mL(0.089 mol)氯乙醇、15 mL三乙胺加入到250 mL烧瓶中,加热搅拌回流12 h,降至室温,抽滤,用少量甲苯洗涤,再依次用稀盐酸(2×40 mL)、饱和食盐水(3×40 mL)、水(2×40 mL)洗涤,有机层加无水Na2SO4干燥过夜,滤去Na2SO4,蒸去甲苯,即得化合物Ⅴ。
(5)左旋西替利嗪二盐酸盐(Ⅵ)的合成
取70 mL经干燥处理的叔丁醇加入到150 mL三口烧瓶中,加入8.0 g(0.051 mol)叔丁醇钾,搅拌让其完全反应形成均匀溶液,加入9.5 g(0.028 mol)化合物Ⅴ,加热搅拌回流1 h;然后加入3.5 g(0.029 mol)氯乙酸钠,回流2 h,再加入1.8 g(0.019 mol)氯乙酸钠;1 h后再加入0.90 g(0.009 mol)氯乙酸钠,反应10 h;停止加热,减压蒸去叔丁醇;残留物加100 mL水,于50 ℃搅拌数分钟,加36%盐酸约28 mL调pH值至10~12;用乙醚(3×30 mL)萃取,分出水相;加36%盐酸约25 mL调pH值约为2~3;用二氯甲烷(2×30 mL)萃取,合并二氯甲烷层,用饱和食盐水(2×30 mL)洗涤,无水Na2SO4干燥,浓缩,向残留物中加入94 mL乙醇、1.4 g活性炭,加热回流1.5 h,冷至室温,过滤,减压蒸去乙醇,得到微黄色透明粘稠油状物约6.3 g,取33 mL36%盐酸,搅拌使之溶解,减压蒸干,得到微黄色固体,将其加入到68 mL丁酮中,搅拌加热回流3 h,冷至室温,抽滤,滤饼用丁酮(2×13 mL)洗涤,于50 ℃真空干燥4 h,得目标化合物Ⅵ。
1.2.2N,N-二氯乙基-4-甲氧基苯磺酰胺的合成
N,N-二氯乙基-4-甲氧基苯磺酰胺无市售产品,因此,本实验以苯甲醚与氯磺酸合成了甲氧基苯磺酰氯,再与双氯乙基胺盐酸盐反应制备了N,N-二氯乙基-4-甲氧基苯磺酰胺,总收率67.4%。
(1)甲氧基苯磺酰氯的合成
取苯甲醚81 mL溶于300 mL CHCl3中,冰浴降至-8 ℃;取98 mL氯磺酸滴加到上述配好的溶液中,保持溶液温度低于0 ℃,在0.5 h内滴加完毕;移去冰浴,升温至20 ℃左右,继续搅拌约1 h后,倒入冰水中,用 150 mL氯仿萃取2次;减压蒸去大部分氯仿,留少量液体冷冻结晶后抽滤,滤饼真空干燥,即得甲氧基苯磺酰氯。
(2)N,N-二氯乙基-4-甲氧基苯磺酰胺的合成
取双氯乙基胺盐酸盐89.25 g(0.5 mol)、CH2Cl2650 mL、三乙胺145 mL,置于反应瓶中;1 h内将其慢慢滴加到300 mL含95 g甲氧基苯磺酰氯的CH2Cl2溶液中,滴加完毕,升温至40~43 ℃,反应6 h,蒸除溶剂,即得N,N-二氯乙基-4-甲氧基苯磺酰胺。
2 结果与讨论
2.1 中间体及目标化合物的结构表征

化合物Ⅲ:白色固体,16.8 g,收率85.2%。1HNMR(CDCl3,300 MHz),δ:2.47(t,4H,3位-CH2CH2-),3.01(t,4H,2位-CH2CH2-),3.92~4.22(m,3H,4位-OCH3),4.22(s,1H,1位),7.72~7.03(m,13H,ArH)。IR(KBr),ν,cm-1:3030(m,Ar的C-H),2968(m,C-H),1594、1493(m,Ar骨架),1452(s,N-CH2),1341(w,CH-),1262~1022(s,CH3O-),756(m,C-Cl)。
化合物Ⅳ:白色固体,5.8 g,收率88.5%,纯度95.9%。1HNMR(CDCl3,300 MHz),δ:2.36(s,4H,2位-CH2CH2-),2.90(s,4H,3位-CH2CH2-),4.21(s,1H,1位),7.38~7.20(m,9H,ArH),1.609(水峰)。IR(KBr),ν,cm-1:3303(m,N-H),3067(m,Ar-H),2953~2749(m,CH2),1596、1486(s,Ar骨架),1181~1005(s,C-H面内),841~695(w,C-H面外),750~715(m,Ar-H相邻)。
化合物Ⅴ:浅黄色透明粘稠物,13.8 g,收率89%,纯度99.3%(文献值[3]:收率90.5%,纯度97.8%)。1HNMR[8](CDCl3,300 MHz),δ:2.28~2.37(s,4H,2位-CH2CH2-),2.43(s,4H,3位-CH2CH2-),2.56(m,2H,4位),3.60(m,2H,5位),4.22(s,1H,1位),7.37~7.21(m,9H,ArH),1.609(水峰)。

2.2 N,N-二氯乙基-4-甲氧基苯磺酰胺的结构表征
甲氧基苯磺酰氯:白色固体,125.1 g,收率81.8%,m.p.39~42 ℃。
N,N-二氯乙基-4-甲氧基苯磺酰胺:微黄色固体,127 g,收率82.4%。1HNMR(CDCl3,300 MHz),δ:2.47(s,4H,5位-CH2CH2-),3.01(s,4H,4位-CH2CH2-),3.92~4.22(m,3H,3位-OCH3),7.72~7.06(m,4H,1,2位ArH-2,3,5,6)。IR(KBr),ν,cm-1:2968(m,C-H),1594、1493(Ar骨架),1452(m,N-CH2-),1341(s,C-N),1262~1022(m,CH3O-),833~656(C-H面外),756(m,Ar-H相邻),756~620(m,C-Cl)。
3 结论
以混旋4-氯二苯甲胺为原料,经化学拆分制得左旋4-氯二苯甲胺(Ⅱ),进而制备左旋4-氯二苯甲基哌嗪(Ⅳ)、左旋4-氯二苯甲基哌嗪乙醇(Ⅴ)和目标化合物左旋西替利嗪二盐酸盐(Ⅵ),并对中间体及目标化合物进行了结构表征。结果表明,化合物Ⅱ收率为33.2%,纯度为98.85%;化合物Ⅳ收率为88.5%,纯度为95.9%;化合物Ⅴ收率为89%,纯度为99.3%;化合物Ⅵ收率为73.5%,纯度为99.1%;总收率为19.08%。其中,采用N,N-二氯乙基-4-甲氧基苯磺酰胺与化合物Ⅱ反应,形成哌嗪环后去保护基所生成的化合物Ⅳ收率高、纯度好。该方法具有操作简便、反应条件温和、反应时间短、对环境友好等优点。
参考文献:
[1] Corey E J,Helal C J.Catalytic enantioselective synthesis of the second generation histamine antagonist cetirizine hydrochloride[J].Tetrahedron Letters,1996,37(28):4837-4840.
[2] 王玉玲,高乾善,周伟,等.左旋西替利嗪盐酸盐的合成[J].食品与药品,2005,7(9):33-35.
[3] Rajan S T,Rao U V B,Prabhakar A S,et al.Processes for preparing levocetirizine and pharmaceutically acceptable salts thereof [P].WO 2009 062 036,2009-05-14.
[4] 王立升,王天文,朱红元,等.左西替利嗪的合成[J].广西大学学报(自然科学版),2007,32(4):384-386.
[5] 刘庆彬,张福平,张占辉.左旋西替利嗪盐酸盐的一种合成方法[P].CN 101 100 462,2008-01-09.
[6] 刘补娥.盐酸西替利嗪合成工艺的改进[J].广东化工,2008,35(9):66-67.
[7] 邹志红,王其远,王伟.左旋盐酸西替利嗪的制备研究进展[J].东南大学学报(医学版),2009,28(4):352-357.
[8] 李茜,沈文斌,邹巧根.盐酸西替利嗪的NMR研究[J].药学学报,2003,38(10):767-770.
[9] 王其远,邹志红.左旋盐酸西替利嗪的合成工艺改进[J].中国药物化学杂志,2008,18(4):273-275.
