X射线显微镜
2012-07-26马礼敦复旦大学分析测试中心同步辐射研究中心
马礼敦/复旦大学 分析测试中心、同步辐射研究中心
0 引言
X射线显微镜是X射线成像术的一种,也是显微成像技术,即将微观的、肉眼无法分辨看出的结构、图形放大成像以便观察研究的器械。在《上海计量测试》2010(5,6)期的《X射线成像术》[1,2]一文中已介绍X射线成像的衬度原理、设备的构造与主要组成部件(如X射线源、探测器等),但主要是从宏观物体的成像(如人体器管的医学成像、机械制品的缺陷探伤、机场车站的安全检查等)出发的。宏观成像与微观成像有相通之处,如衬度原理、设备的主要组成部件等,但也有区别。由于X射线显微镜是用来观察肉眼无法分辨的微观结构与图形,因而在仪器结构和要求上有显著不同,如要求光源尺寸小而强度大,要将像放大和高分辨等。本文主要叙述不同之处。
1 X射线显微镜的成像与构造
X射线显微镜的成像原理与光学显微镜基本上是一样的,遵从几何光学原理,其关键部件是成像和放大作用的光学元件,在光学显微镜中为透镜。由于X射线的波长很短,在玻璃和一般物质界面上的折射率均接近1,故其成像放大元件不能用玻璃透镜,现在一般用波带片。此外,它们同样利用吸收衬度和位相衬度成像,同样要求有强光源及像探测器。对光学显微镜,一般用肉眼观察,故常加一目镜起进一步放大的作用。在X射线显微镜中当然不能用眼晴直接观察,可用CCD等面探测器探测。显微镜的重要性能指标两者是相似的,有放大倍数、分辨力、像差等几个。X射线显微镜的一般构造见图1。从强光源来的光束先经聚焦元件(在此为毛细管透镜聚焦)使光斑尺寸变小、亮度加大,然后射到样品上,透过样品的光,再经成像放大元件(在此为波带片)而到达探测器(在此为闪烁体加CCD)。成像波带片和探测器之间有一个Au位相补偿环,在相衬成像时用。如吸收衬度成像,可移走。关于衬度,参考文献[1]中已有提及,下文将对聚焦放大元件、光源和探测器几个问题进行讨论。
1.1 聚焦放大元件[3]
常用的聚焦镜是多层膜反射聚焦镜和波带片,成像放大元件是波带片。

图1 X射线显微镜的构造
1.1.1 多层膜反射聚焦镜
多层膜是在基板上重复涂上两种不同的材料制成的人造一维晶体。通常,一种材料是高原子序数的重金属(H),另一种是低原子序数的非金属(L)。这两个层的厚度之和dH+dL构成这多层膜的重复周期d。dH和dL的大小与其比值及多层膜的性质有关。需按实验的要求来设计、制造,因此品种很多。最简单的是膜厚d均匀的镜子,但也有膜厚d是逐渐变化的镜子。镜面可以是曲面,可以是一维弯曲的(如圆筒面、抛物面或椭圆面等),也可以二维弯曲的(如球面或椭球面等)。图2是多层膜椭圆面反射聚焦镜的构造示意图。发散光源放在椭圆的一个焦点上,反射光聚焦在另一个焦点上。

图2 多层膜椭圆面聚焦镜的原理
这种一维弯曲的聚焦镜只能在一个平面内起聚焦作用,得到的是线焦点,与此平面垂直的方向,射线仍保持原发散情况。若要两个方向都聚焦获得焦点可用两种方法:一是在两个方向上做二维弯曲;另一是将两块聚焦镜转过90°先后放置,构成所谓的Kikpatrick-Baez (K-B)系统(图3)。K-B系统有较多的实际应用。

图3 K-B系统示意图
1.1.2 Fresnel 波带片
Fresnel波带片的原理早在100多年前就已提出,但将其用于X射线却是近十几年的事。波带片的常见形状见图4。是由两套不同材料制成的同心圆环交替排列构成的,其中一种材料透光,另一种不透光。设入射光是波长为λ的平面单色光,焦距为f,设计焦点使到各相邻环带的距离均相差λ/2,则通过两个相邻透明环带到达焦点的这两组光程差为λ,具有相同的位相,干涉相会长。焦距为f的波带片可以有不止一个的焦点,这些焦点f1、f2、…到各相邻环带的距离差分别为 3λ/2、5λ/2、…,其焦距分别为f/ 3、f/ 5、…,为不同级次的聚焦。

图4 波带片构造示意图
波带片中各环带的半径R近似为式中k为中心往外数的环带顺序号,f为波带片焦距[f= 4N(Δr)2/λ,N为环带的总数]。波带片各环带的宽度是向外逐渐变小的,最外一个带的带宽为Δr。Δr决定了波带片成像的衍射限分辨力δ(δ= 1.22Δr)。
上述的波带片称振幅波带片,其效率较低。为了提高效率,要将不透光的部分也变成透光,同时设法使它与透过相邻环带的光线间的光程差不是λ/2,而是λ,相位从π变到2 π,成为同相位,干涉相就长了。方法可以是涂膜或使用两种有不同折射率n的材料来制作波带片,使光在通过波带片后附加了一个相位差π。这种波带片称相位波带片。理想的相位波带片,对一级聚焦,η可达40%,比振幅波带片提高了几倍。图5为金制作的波带片的效率随金厚度及X射线光子能量变化的曲线[3]。

图5 金制波带片的效率随金厚度及X射线光子能量变化的曲线
1.1.3 毛细管
毛细管的聚焦作用是基于X射线在毛细管内壁上的全反射,来改变X射线的前进方向实现的。毛细管若是弯曲的,则会引导X射线改变方向。毛细管不是等径而是锥状的,则X射线就会在靠近出口处聚焦[图6(a)]。它既可以聚焦单色X射线,也可以聚焦白色X射线。可聚焦的X射线能量范围约从200 eV至80 keV。
利用单根毛细管聚焦X射线束的实物在20世纪80年代已被实际应用。美国康奈尔大学用硼硅玻璃做的一根毛细管,长为1.6 m,粗头直径为470 μm,细头直径为110 μm。此管的反射效率对8 keV的射线是49%,对13 keV的射线为54%,对20 keV的射线为34%。反射效率低于100%是因为毛细管的表面并不平整,一定的起伏和粗糙度会损失部分射线。从单根毛细管射出的光束的直径在出口处为最小,以后会慢慢增加。若毛细管出口直径为r,则工作距离为20 μm至100 μm,此值是比较小的。
Heald等研究过毛细管的长度、进出口直径等参数与毛细管的透过率、强度增益倍数等性能的关系,部分结果见表1。

表1 毛细管结构参数与性能参数
毛细管除了单根利用外,还可以集束应用。毛细管束,就是将数千根毛细管平行聚集在一起的元件,整体呈圆锥状,图6(b)为其示意图。一个由336根直径为17 μm的玻璃毛细管组成的毛细管簇,可使光斑面积缩小至约1/20,功率密度可提高5倍。
从图6可以看到,若入射光从毛细管锥体的大头入射,则从小头射出的光被聚焦;如入射光从较小的一头进去,则从较大的一头射出的光为平行光。与单毛细管相比,毛细管束的长度可以短,其焦点的面积会比单毛细管大一些,但焦点的位置离管口的距离也会大一些(有几公分),使用起来比较方便。毛细管束还可以是中间大两头小的双圆锥形,见图6(c)。
聚焦元件还有用单晶体做的和所谓的折射透镜,不过,它们在X射线显微镜中用得不多。
1.2 光源
[1]中已介绍了三类X射线光源:实验室X射线光源(X射线管)、直线加速器和同步辐射装置。同步辐射是既近平行又高强度,且波长可调而成为最理想的光源。未见有将直线加速器用于X射线显微镜,实验室光源有使用的,但不能用焦点在10 mm×1 mm左右的封闭X射线管,可以用高功率的旋转阳极X射线管[3,4]。另外,可用焦点尺寸在数十微米的细聚焦X射线管[4],该管焦点小,但光亮度大,可达 2×1011~1.2×1012photos/s•sr,已接近同步辐射的光亮度,而且功耗很小,一般只有数十瓦,整机体积也小,适合作为实验室X射线显微镜的光源。此外,还有用等离子X射线源[4,5]的。从光源发射出的X射线一般需先经聚焦使光斑进一步缩小,光亮度进一步增加才照到样品上。总的来说,实验室光源的光亮度比较低,仪器的分辨力也低,故实验室X射线显微镜应用面不广,而同步辐射显微镜却发展得很快,做出了许多很有价值的工作。

图6 (a) 单根毛细管 (b) 毛细管束 (c) 双圆锥形毛细管束
1.3 探测器
参考文献[1]中已介绍的各种探测器都可用,如感光胶片、影像板(Image plate, IP)、影像增强器、半导体探测器(CCD,电荷偶合器)等。当然,宏观用的和微观用的在结构和参数上是不同的。
X射线显微镜可按使用的X射线能量的高低分为软X射线显微镜和硬X射线显微镜。其构造基本相同,但研究对象有侧重。前者较多应用于生物医学体系,后者较多应用于材料医学体系。如按其技术或原理来分类,则有透射式或扫描式显微镜、光谱显微镜、全息显微镜等多种。
2 透射式X射线显微镜[6]
图7为透射式X射线显微镜的构造示意图。用波带片作为聚光镜、显微波带片作为成像放大物镜、CCD为探测器,分辨力可达10 nm。将样品连上了制冷装置(氦气)、转动机构,并使CCD与计算机连接,则可做断层扫描(CT),并从屏幕上直接观察CT图。
水窗:水窗是指从波长2.3 nm至4.4 nm的一个波段范围。用此范围的X射线入射于生物样品,水中含有的氧对它们的吸收很小(<40%),而有机体中的碳对它们的吸收却很高(>60%)(见图8),可以认为水对此波段的光是透明的,使水和氧间的衬度很高。故用此波段的光来观察含水生物样品,可避开氧吸收的干扰,清楚看到主要由碳构成的有机分子的构造。图9为用波长为2.4 nm的X射线拍摄的冰冻Kupffer 细胞图像,除细胞核N外、可清晰分辨出细胞核膜Me和囊泡V等[7]。
透射式X射线显微镜既可利用吸收衬度成像也可用相位衬度成像。这两类仪器在构造上略有差别,图10为相衬显微镜构造示意图[8]。相衬显微镜的聚光器不是单一的波带片,而是由环状孔径、波带片和针孔构成。环状孔径将入射光限制为一个环,波带片将其单色化并聚焦在位于针孔中的试样上,从试样出射的光经显微波带片和位于其背焦面附近的环状相位板(添加位相)而成像在探测器上。

图7 透射式X射线显微镜光路示意图
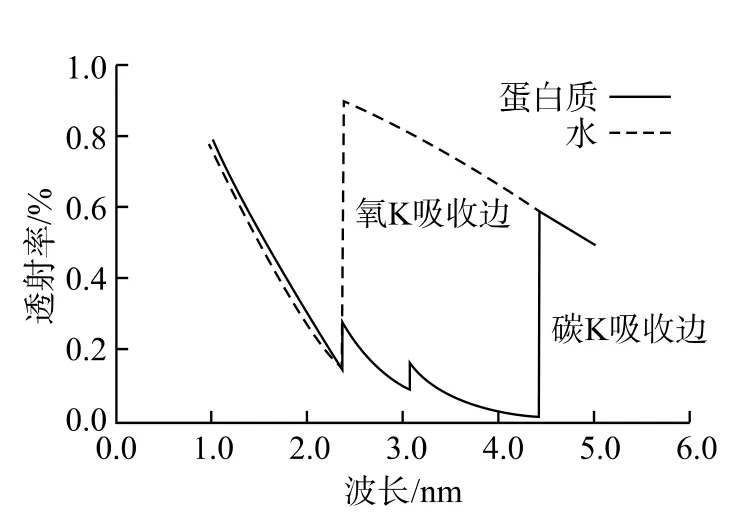
图8 氧和碳的吸收限和水窗

图9 Kupffer 细胞的X射线显微照片

图10 相衬X射线显微镜光路
图11是脂质囊胞膜的X射线显微照片[8],(a)为相位衬度像,(b)为吸收衬度像,显然,前者比后者能更清楚地看到结构细节。

图11 脂质囊胞膜的X射线显微照片
3 扫描式X射线显微镜[6]
在上述透射X射线显微镜中,整个被研究物需完全暴露在入射光束中,探测器显示的是放大、完整的物像。在扫描式X射线显微镜中入射光束一般被聚焦得很细小,如几十个纳米,故物体上只有一个很小的区域被光照射,探测器上只得到这一个点的放大图像,相对移动物体与光的位置,可逐点得到物体上各点的像,这些点像被逐点输入计算机,经处理后在显示器上显示出完整的图像。图12为扫描式X射线显微镜构造示意图[9]。一般用扫描台实现,扫描步距可以为几个纳米。可以是光路不动,样品移动逐点扫描,也可以样品不动波带片扫描(见图12)。一般是样品移动的分辨力较低,波带片扫描的分辨力较高。聚光波带片之后常有一个级次选择光阑(OSA),用来从波带片产生的不同级次的衍射光中选择需要的级次,并阻挡其他级次的光,以降低背景。

图12 扫描式X射线显微镜构造示意图
扫描式X射线显微镜可以利用吸收衬度成像,也可用相位衬度成像;可得到明场像,也可得到暗场像。
4 光谱显微镜[10]
所谓光谱显微镜是将某种光谱和显微镜相结合的技术。此类技术颇多,只要这种光谱能产生一种与观察位置有关的信号,如透射光子的计数率、总的或特定的峰的光电子产率、荧光产率等,这些信号可以给出元素的、化学的、磁的等各种信息。图13为此类仪器光路示意图,入射X射线经两次多层膜反射聚焦到试样上,样品可在两个方向扫描,以得到整个图像。探测器依据需探测的信号的性质选择品种、安装位置等。如欲探测的是发生的多色荧光,则可用固体探测器在反射位置探测;如探测的是单色的透射或衍射线,则可用闪烁计数器或CCD等测定;如被探测的是光电子,则需用电子分析器探测。
4.1 NEXAFS显微镜

图13 光谱显微镜构造示意图
这是将X射线吸收光谱[11]与X射线显微镜结合的技术。NEXAFS是X射线吸收光谱中靠近吸收边的那一段光谱。对一些有机或生物分子,如核酸与蛋白质、聚苯乙烯PS和有机玻璃PMMA或PEP等,由于它们均由C、H、O等轻元素构成,利用一般的吸收衬度是不能区别的。但如选择某种特定波长的X射线,则可将它们区分出来。这是因为在不同化合物中的C、H、O有着不同的近邻配位结构,因而它们的外层能级结构不同,造成它们的X射线近边吸收光谱不同,故可比较它们的NEXAFS谱,找出它们的吸收有巨大差异的能量,用这种能量的X射线入射,则一种物质吸收小而另一种吸收大,可以造成大的吸收衬度,明显地将它们加以区别。图14右下图为三元高聚物中三个组分PS、PMMA和PEP的NEXAFS谱。可看出,在入射线能量为285.15处,PS吸收很大,另两个几乎不吸收;在能量为287.6处,PEP吸收大,另两者吸收小;而在能量288.4处是PMMA吸收很大,而PS和PEP吸收很小。分别用这三个能量的单色X射线做扫描X射线显微镜观察,所得显微图像分别位于图14的左上、右上和左下。这三个图的暗处分别表示了PS、PEP和PMMA的存在。从这些显微图像上可分辨出三个组份各自的形态和相互之间关系。此三元聚合物中的大颗粒主要由PS构成,PS中存在许多小颗粒,是为PMMA,而大颗粒之间充填的是PEP[12]。

图14 三元聚合物的光谱显微图像和谱图
4.2 磁X射线显微镜
同步辐射中所含的辐射均是偏振光,可以是线偏振光,也可以是椭圆或圆偏振光,X射线也不例外。如果待测物质具有磁性,则具有不成对电子,具有电子自旋磁矩和轨道磁矩。磁矩与不同方向的偏振光的作用是不同的,如用不同方向的圆(线)偏振光照射磁性材料,可以得到不同的吸收谱,该性质称圆(线)二色性。把用左右圆(线)偏振光得到的不同的吸收谱相减就得到圆(线)二色谱。图15是德国BESSYⅡ的磁X射线显微镜的示意图[13]。X射线源来自一同步辐射装置中的椭圆波荡器,经前置镜使其准直,滤波后经一离轴波带片聚焦,再经单色器(由三块平晶组成)使其成为单色光,并聚焦到试样上,经试样吸收后的光用一显微波带片成像并放大,像用CCD探测。在试样台上装有可以施加不同方向外磁场的系统。
4.3 光电子发射显微镜[10]
光电子发射显微镜是利用X射线在样品上激发出光电子来放大成像的装置。如要研究磁性材料,则和磁显微镜一样,入射光需偏振X光。图16右为光电子发射显微镜构造示意图[14],与电子显微镜类似,由几个电磁透镜起成像放大作用。图16左下为样品磁畴构成示意图,由四个不同磁矩方向(用箭头表示)的磁畴构成,入射圆偏振X射线与不同磁畴的作用不同,激发出光电子不同,故所成之像不同,可区别出4个磁畴(图16左上)。

图15 磁X射线透射显微镜

图16 X射线光电子发射显微镜
图17[14]上图为生长于Cu(100)面上的Co (4ML)/Cu (6ML)/Ni(15ML)三层膜(Cu层没画出,Co层在上)示意图。用波长与Co L3吸收边的波长相同的X射线照射,Co层强烈吸收,Ni层吸收少,获得Co层的磁畴像,示于图17下右。颜色不同的区域表示磁化方向不同,上图Co层内的两个箭头表示磁化方向,是在平面内的;换用与Ni L3吸收边能量相同的X射线入射,则得到的是Ni层的磁畴像,见图17下左。磁畴呈条状平行排列,但磁化方向不在平面内,而是与平面垂直,示于上图。Ni、Co层的磁畴形状不同,但Co层的结构似与Ni层的条状结构有一些关系。

图17 Co(4ML)/Cu(6ML)/Ni(15ML)三层膜层分辨磁畴像
图18(a)是利用线偏振光的光路示意图。线偏振光以与样品表面小于15°角的方向入射,若是s偏振,则电矢量与样品表面平行;若是p偏振,则电矢量与样品表面法线成15°角。图18(b)为利用能量位于Ni L吸收边的线偏振光拍摄得到的Ni(100)表面上的反铁磁畴的光电子发射像,呈现出Ni表面条状的反铁磁畴结构[15]。
5 X射线全息显微术[6]
已经知道,像是依靠吸收衬度(光的振幅)或位相衬度一种信息来显现的。而所谓全息,是指同时含有振幅与位相两种信息。这是Gabor在1948年提出的。由于目前的记录介质实际可记录的信息只能是光强,也即振幅,故需将位相信息转换成强度来记录。把光照射到试样上,试样以球面波形式将其散射,如有另一束已知振幅与位相的、未经散射的直射参考光,两者的波前相遇时因经过的途程长度l与l0不同,存在位相差,故会干涉,则合成的总强度是既与原光的振幅有关,又与两光的位相差有关。故此总强度包含了振幅与位相两种信息[图19(a)]。这样的干涉图是全息图,并不是原物体的像,怎样才可从干涉图获得原物体的图像呢?需通过图像再现。这就要用激光来照射记录下的干涉图,其发出的波前仍和参考源相同,但其干涉图又与试样的波前一致,从而再现原物体。全息显微过程是由全息记录和波前再现两步构成的。

图18 线偏振光的(a)光路、(b)磁畴像

图19 X射线全息显微原理图
X射线荧光全息(XFH)[16]已被用来获取局域原子的结构。在荧光全息中,入射X射线激发试样中感兴趣元素的荧光。荧光的一部分直接射向探测器,另一部分被其周围的原子散射,散射荧光射向探测器,与直射荧光发生干涉而得到这一方向k的全息强度I(k)[图19(b)]。在一个相当大的立体角范围内改变探测器的位置,也即改变方向,改变l、l0的长度,改变了位相差,则每一方向得到的干涉强度是不同的,其全体即为全息谱。
荧光全息还有另一种实现方法,为多能X射线全息(MEXH),其光路是XFH的倒转,探测器和激发光源互换位置[图19(c)]。入射光一部分被近邻原子散射,散射波到荧光发生原子处与直接到达的入射光发生干涉,荧光发生原子接受到的光强是上两光的干涉结果,故该原子发射的荧光强度是与干涉结果相关联的。与XFH类似,改变入射光的位置,以得到不同方向的I(k),加合得到全息图。改变入射光的波长,可得不同波长的全息图,加大了倒易空间中的采样范围,避免像的畸变。加和这些全息图还可有效解决孪生像的问题。Marchesini等用XFH法研究了准晶Al70.4Pd21Mn8.6的结构[17]。实验是在欧洲同步辐射装置ID22波荡器光束线上进行,入射光能量为16 keV。图20(a)为实验全息图,经积分变换得到的实空间的原子结构见图20(b)。联接其中最亮的12个点可看出准晶的五角二十面体特征。

图20 Al70.4Pd21Mn8.6的(a)XFH全息图、(b)原子结构图
6 X射线显微镜与其他显微镜的比较
表2中列出了X射线显微镜与其他显微镜几项主要性能的比较。可看出各种显微镜均有自已的特色及适用范围。分辨力最高的是电子显微镜,其次是X射线显微镜。由于电子的穿透力非常低,故电子显微镜适用在表面形貌和结构的分析,而X射线有强的穿透力,故可做块体分析,甚至做断层扫描(CT)。X射线显微镜的另一特点是可与各种光谱结合成光谱显微镜,大大扩大了它的应用范围。

表2 X射线显微镜与其他显微镜比较
参考文献:
[1] 马礼敦. X射线成像术(上)[J]. 上海计量测试. 2010(5). 2-10.
[2] 马礼敦. X射线成像术(上)[J]. 上海计量测试. 2010(6). 2-12.
[3] 马礼敦. 近代X射线多晶体衍射——实验技术与数据分析[M]. 北京:化学工业出版社, 2004. 242-272.
[4] 马礼敦. 近代X射线多晶体衍射——实验技术与数据分析[M]. 北京:化学工业出版社, 2004. 92-109.
[5] J. Thieme,B., Nieman,G. Schmall. U.S.P 5222113[P]. 1993.
[6] 金承志. 第九章 软X射线显微术[M]. //马礼敦,杨福家. 同步辐射应用概论, 第2版. 上海:复旦大学出版社, 2006. 454-520.
[7] Scharf J, Schneider G. Ultrastructural characterization of isolated rat kupffel celles by transmission X-ray microscopy[M]. // J of Microscopy, 1999, 193:250.
[8] Schmahl G, Rudolph D, Guttmann P, et al. Phase contrast X-ray microscopy[M]. // Synchrotron Rad News, 1994, 7(4):19-22.
[9] Wiesemann U, Thieme J, Guttmann P, et al. First results of the new scanning transmission X-ray microscope at BESSY Ⅱ[M]. // J Phys Ⅳ,2003, 104:95.
[10] Advanced Light Source. X-ray Spectromicroscopy. ALS 1997, 35.
[11] 马礼敦, 施国顺. 第七章 X射线吸收精细结构光谱[M]. // 马礼敦.高等结构分析. 上海:复旦大学出版社. 2006. 323-374;
[12] Ade H, Hitchcock A P, Mitchell G E, et al. Applications with the dedicated polymer scanning transmission X-ray microscope at ALS beamline 5.3.2[J]. Synchrotron Radiation News. 2003, 16(3): 53-59.
[13] Eimuller T, Niemann B, Guttmann P, et al. The magnetic transmission X-ray microscopy project at BESSY Ⅱ[J]. J Phys Ⅳ, 2003, 104:91.
[14] Kuch W, Chelaru L I, Offi F, et al. Layer-resolved magnetic domain imaging using X-ray photoelectron emission microscopy[J].Synchrotron Radiation News. 2002, 15(6):12-16.
[15] Ohldag H, Weber N, Bethke C, et al. Imaging of antiferromagnetic domains by linear magnetic dichroism in photoemissionmicroscopy of NiO(100)[J]. Synchrotron Rad News, 2000,13(6):25-32.
[16] Len P M, Gog T, Fedley C S, et al. X-ray fluorescence holography and multi-energy X-ray holography: A critical comparison of atomic images[J]. Phys Rev B, 1997, 55(6): R3323-R3327.
[17] Marchesini S, Shimithusen F, Tegze M, et al. Direct 3D imaging of Ai70.4Pd21Mn8.6 quasicrystal local atomic structure by X-ray holography[J]. Phys Rev Lett, 2000, 85(22): 4723-4726.
