锰(Ⅱ)咪唑二羧酸基超分子的水热合成、晶体结构和热性能
2012-07-12李晓春
李晓春
(赤峰学院化学系,内蒙古赤峰 024001)
最近,随着测试技术以及合成技术的进步,配合物功能材料迎来了新的发展机遇。以4,5-咪唑二羧酸及其衍生物为基础配体通过分子自组装构筑的新型功能配合物在新材料应用、离子交换和分子识别等方面有广阔的发展前景。其中,含2位取代基咪唑双羧酸具有4,5-咪唑二羧酸配体的优点,它们同样因有配位能力强的氮原子和氧原子而能够和不同的金属离子形成多种多样的配位化合物。该类配体可以以单齿、双齿和多齿的配位模式与金属离子连接起来而形成特殊复杂有趣的配位化合物[1]。截止目前,含2位取代基的咪唑双羧酸类配聚物文献中已有一些报道,但是总体来说数量仍然有限。尽管文献中已有不少由2位脂肪取代基形成的配合物,然而,2位芳香基取代的咪唑羧酸类配合物仍然为数不多[2],这为我们研究芳香基团修饰的咪唑羧酸类配合物提供了广阔的空间。
此外,在构筑结构复杂的配位化合物的过程中,芳香羧酸配体以及有机含氮配体都是很重要的有机配体。人们希望利用羧酸配体的配位形式的多变以及含氮配体的强配位能力,去合成一些结构新颖、性能独特的功能配合物。
在本文,我们报道了一个2-(-萘基)-咪唑-4,5-二羧酸(H3NIDC),通过水热合成所构筑的一个结构新颖的锰(Ⅱ)的超分子配合物:[Mn(H2NIDC)2(H2O)2]的合成、晶体结构,并研究了其在空气气氛下的热稳定性能。
1 实验部分
1.1 试剂及仪器
试剂:所用试剂均为分析纯品,使用前未进一步纯化。有机配体H3NIDC参照文献方法合成[3]。
仪器:元素分析使用美国FLASH 1112元素分析仪,红外光谱在400~4 000 cm-1范围内用KBr压片在美国Nicolet NEXUS 470-FTIR红外光谱仪上测定。晶体结构利用Bruker Smart 1000 CMN衍射仪测定。样品的热分析在空气气氛下,以10℃/min的升温速率采用NETZSCH STA 409PC同步热分析仪测定。
1.2 配合物[Mn(H2NIDC)2(H2O)2]的合成
将 H3NIDC(0.014 2 g,0.05 mmol)、MnCl24H2O(0.004 9 g,0.025 mmol)混合,然后加入 8 mL蒸馏水和甲醇的混合溶液(1∶1),搅拌均匀,把混合液放入聚四氟乙烯内衬中,再放入不锈钢反应釜中,密封,放入烘箱中升温至150℃,恒温96 h后,以10℃/h的速率降温。缓慢降至室温后,有淡黄色的块状单晶生成,该晶体在溶液中稳定。产率:62% 。IR(cm-1,KBr):3565 s,3096 s,1708 s,1 648 w,1 530 s,1 400 m,1 271 w,1 228 w,1 102 w,994 w,920 w,820 w,758 m,725 w,634 m,540 m。
1.3 衍射数据的收集和晶体结构测定
选取大小为1.85 mm ×0.21 mm ×0.20 mm 的无色单晶,296(2)K下在Bruker Smart 1000 CMN衍射仪上,用石墨单色器单色化的Mo-k射线(λ=0.710 73 nm)收集到独立的衍射点有10 161个,其中Ⅰ>2σ(Ⅰ)的点为2 550个。结构分析表明,晶体属单斜晶系,空间群为P2(1)/c,所得晶胞参数为:a=0.839 30(14)nm,b=0.587 49(10)nm,c=2.6969(5)nm,α =90,β =93.021(2),γ =90,V=1.3279(4)nm3,Z=2,Dc=1.629mg/m3,μ =0.569 mm-1,F(000)=666。衍射数据经 Lp因子校正后,采用直接法初步解出各原子位置坐标,继而经差值Fourier合成及最小二乘法修正和各向异性温度因子校正,结构参数211个。最终偏离因子R=0.040 8,Rw=0.099 0。最终残余电子密度最高峰ΔPmax=356 e/nm3,最低峰 ΔPmin=-270 e/nm3,所有的计算均使用SHELXL-97程序包[4]。
2 结果与讨论
2.1 晶体结构分析
标题配合物的部分键长和键角数据列于表1、表2中。

表1 标题配合物的部分键长 nm

表2 标题配合物的部分键角(°)
[Mn(H2NIDC)2(H2O)2]是一个单核配合物,中心Mn(Ⅱ)离子分别于来自于两个H2NIDC-阴离子上的两个羧基氧原子、两个咪唑氮原子以及两个配位水上的两个氧原子形成了六配位。Mn(1)-O(2)以及Mn(1)-O(5)的键长分别为0.214 85(19)nm 和0.220 0(2)nm;Mn(1)-N(1)的键长为0.231 6(2)nm。键角O(2)-Mn(1)-O(2A)、O(5)-Mn(1)-O(5A)和N(1)-Mn(1)-N(1A)都为 180.0°,其它键角在 75.96(7) ~ 104.04(7)°之间。O(2)、O(2A)、O(5)、O(5A)、Mn(1)完全位于同一平面,两个配位水分子处于轴向位置,由这些数据可以看出该八面体构型接近理想的正八面体,仅有稍微的变形(图1)。
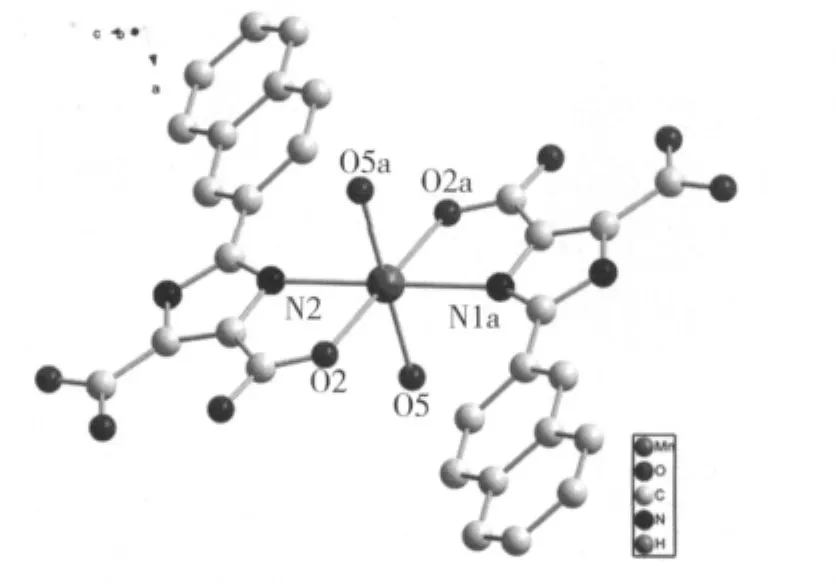
图1 标题配合物的结构单元图
需要指出的是,两个咪唑二羧酸配体都是只脱去了一个羧基氢原子,形成了负一价的阴离子。在同一个有机配体中,咪唑环(N1-C13)的原子平均偏离值为0.000 55 nm,萘环(C2-C11)的原子平均偏离值为0.000 87 nm,可见两个平面环的共平面性在参与配位后仍然比较好。两个芳环的二面角为17.0°,显然两环之间由于位阻效应有所扭曲。很明显,不同的有机配体上的两个咪唑环间二面角为0.00°。
从固态结构中可以发现,该配合物中咪唑羧酸配体上的咪唑环和萘环芳香环,它们在固态堆积中,都表现为错位得面对面排列且面与面之间的距离为约0.358 nm,显示芳香环间存在强的π-π堆积作用,从而使配合物结构更加稳定形成了二维固态结构(图2),进一步的这些二维面在分子间作用力以及羧基和配位水所形成的分子间氢键的作用下,形成了三维的超分子结构。
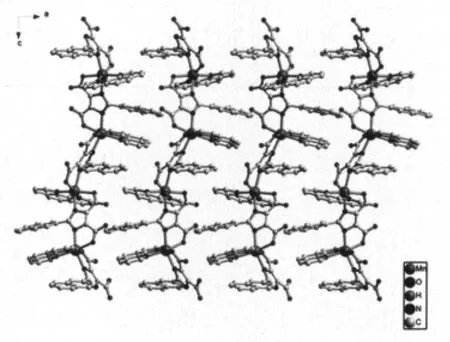
图2 标题配合物通过π-π作用力形成的二维固态堆积图
2.2 光谱分析
此配聚物的红外光谱数据与晶体结构的分析一致。3 565以及3 096 cm-1处的强吸收应为配位水中的O—H伸缩振动,1 708 cm-1和1 648 cm-1处的吸收峰对应配位后羧基不对称伸缩振动,出现在1 400 cm-1和758 cm-1的吸收峰应为萘基上的亚甲基振动。
2.3 热稳定性能分析
图3显示标题配合物在183.5℃之前样品是保持稳定的,它的TG曲线表明该配聚物的失重过程大致可以分为四步:第一步失重是由于两个配位水分子的离去引起的,发生在183.5~222.5℃范围内,实验测得失重比为5.92%(理论值为5.53%);第二步至第四步持续失重从222.5℃开始,直到472℃结束,显示一个持续的失重过程,这部分对应于该结构中两个有机咪唑羧酸配体分子的完全热解(实验值:80.51%)。此后样品残余的质量保持不变。最后剩余的黑色产物推测为MnO2(实验值:13.57%,理论值:13.36%)。
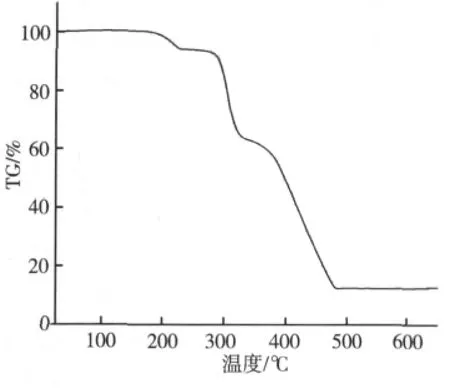
图3 标题配合物的热分解曲线
[1]陈小明.无机合成与制备化学[M].北京:高等教育出版社,2009:363-384.
[2]Li X Wu,B L Niu,C Y,et al.Syntheses of metal-2-(Pyridin-4-yl)-1H-imidazole-4,5-dicarboxylate networks with topological diversity:fas adsorption,thermal stability and fluorescent emission properties[J].Cryst Growth Des,2009,9(8):3423-3431.
[3]Lebedey A V,Lebedeva A B,Sheludyakov V D,et al.Synthesis and N-kylation of 2-lkyl-and-e-arylimidazole-4,5-dicarboxylic acid esters[J].Russ J Cen Chem,2007,77(5):945-953.
[4]Sheldrick G M.SHELX-97,program for the solution and refinement of crystal structures[M].University of Go ttingen,Germany,1997.
