高效液相色谱外标工作曲线法测定大麻树脂中四氢大麻酚含量的不确定度评定*
2012-07-10翟晚枫张春水高利生
翟晚枫 张春水,高利生
(中国人民公安大学刑事科学技术系,北京 100038) (公安部物证鉴定中心,北京 100038)
高效液相色谱外标工作曲线法测定大麻树脂中四氢大麻酚含量的不确定度评定*
翟晚枫 张春水,高利生
(中国人民公安大学刑事科学技术系,北京 100038) (公安部物证鉴定中心,北京 100038)
依据JJF 1059–1999《测量不确定度评定与表示》,以检测大麻树脂中四氢大麻酚含量的实验为例,评定了采用高效液相色谱外标工作曲线法进行含量测定的不确定度。建立了高效色谱外标工作曲线法测定样品含量的数学模型,分析了不确定度的来源并给出了量化结果,合成扩展不确定度为0.15%(k=2)。
高效液相色谱;外标法;不确定度;大麻树脂
测量不确定度是表征合理地赋予被测量值的分散性与测量结果相联系的参数,在测量结果的完整表述中,应包括测量不确定度[1]。当前,在高效液相色谱领域,分析工作者进行不确定度评定的定量方法主要集中于外标单点定量法[2-5]。由于外标工作曲线法测量不确定度的评定较为复杂,因此报道较少[6-7]。外标工作曲线法与单点定量法相比,具有准确、可靠等显著优势,笔者以检测大麻树脂中四氢大麻酚含量实验为例,对采用高效液相色谱外标工作曲线法的测量结果进行了不确定度评定和探讨。
1 实验部分
1.1 主要仪器与试剂
液相色谱仪:Prominenece UFLC型,附带LC-20AD XR型泵、SIL-20AC XR型进样器、SPD-20A型检测器、CTO-20AC 型柱温箱、数据处理软件LC solution,日本岛津公司;
电子分析天平:XP105DR型,感量为0.01 mg,瑞典Mettler公司;
超声振荡器:KQ3200型,昆山市超声仪器有限公司;
台式高速冷冻离心机:德国Sigma公司;
纯水制备系统:Synergy UV型,美国Millipore公司;
移液枪:容量为100~1 000 μL,德国Eppendorf公司;
瓶口移液器:标称容量10 mL,德国Brand公司;
进样针:容量分别为50,100,250 μL,瑞士Hamilton公司;
四氢大麻酚标准品:(100±0.5) μg/mL,美国Cerilliant公司;
大麻树脂样品:案件中收缴;
甲醇:优级纯,国药集团化学试剂有限公司;
乙腈:色谱纯,美国Fisher Scientific公司;
实验用水为去离子水,由蒸馏水经Millipore公司生产的Synergy UV型纯水系统制备而得。
1.2 色谱条件
色谱柱:岛津Inertsil ODS-SP柱(150 mm×4.6 mm,5 μm);流动相:乙腈-水;梯度程序:以乙腈-水(体积比为70∶30)为初始流动相,然后有机相体积分数以2%/min升至96%;柱温:40℃;流速:1 mL/min;进样量:10 μL。
1.3 实验方法
(1)样品前处理
使用玛瑙研钵将样品研磨成粒度均匀的粉末,使用高精度天平等量称取样品粉末6份,分别装入一次性试管中,用瓶口移液器加入20 mL(V1)甲醇,密封后以1 500 r/min振荡10 min,离心5 min,用移液管移取1 mL(V2)上清液装入一次性试管,再用瓶口移液器加入20 mL(V3)甲醇,摇匀后使用移液器移取约1.5 mL溶液装入自动进样瓶中。
(2)四氢大麻酚标准溶液的配制
将四氢大麻酚标准品溶液(0.1 mg/mL)分别用甲醇稀释成浓度分别为c1,c2,c3,c4,c5的标准使用液,供高效液相色谱仪分析(每种浓度进样3次),用最小二乘法建立外标工作曲线。稀释的具体过程如下:
使用注射器将安瓿瓶中1.0 mL质量浓度为0.1 mg/mL的四氢大麻酚标准样品全部吸取后注入容量瓶中,之后使用该注射器吸入稀释溶剂甲醇润洗注射器和安瓿瓶,将剩余有效成分全部转移至容量瓶中,重复多次,定容至10 mL,配制成0.01 mg/mL(c1)的四氢大麻酚标准工作液。
使用标称容量250 μL的微量进样针吸取0.01 mg/mL四氢大麻酚标准工作溶液500 μL(分两次吸取),转移至1 mL单标线容量瓶中,定容,配制成0.005 mg/mL(c2)的四氢大麻酚标准工作液。
分别使用不同标称容量的微量进样针吸取一定体积的0.01 mg/mL四氢大麻酚标准工作液,转移至单标线容量瓶中定容,分别配制成0.002,0.001,0.000 5 mg/mL(c3,c4,c5)的四氢大麻酚标准工作液。
(3)样品测定
将高效液相色谱峰面积响应值分别代入外标工作曲线,得到对应浓度的预估值并分别计算出6份样品的含量,取平均值作为测定结果。
2 不确定度来源分析
高效液相色谱(外标工作曲线)法测定大麻树脂中四氢大麻酚含量的数学模型:

式中:wi——单份样品中四氢大麻酚的质量分数,%;
cpred——由工作曲线计算得到的单份样品溶液的质量浓度,mg/mL;
V1——溶解样品时加入甲醇的体积,mL;
V2——从甲醇一次提取液中移取样品溶液的体积,mL;
V3——稀释时加入甲醇的体积,mL;
mi——单份样品的质量,mg;
n——样品份数。
参照式(1)、式(2),根据JJF 1059-1999[1],高效液相色谱外标法测定大麻树脂中四氢大麻酚含量的测量不确定度主要来源于以下几个方面:采用校准工作曲线计算引入的不确定度(包括不同浓度的响应值A的随机变化、导致系列标准浓度赋值ci误差的随机效应)、用于测定的样品质量m引入的不确定度(来源于天平称量样品的过程)、溶解样品的溶剂体积V1引入不确定度、从甲醇一次提取液中移取V2体积的样品溶液引入的不确定度、稀释时加入的溶剂体积V3引入的不确定度、测量重复性引入的不确定度urep(包括样品不均匀性、技术人员个人熟练程度和其它因素引入的不确定度)。
3 不确定度分量的量化
3.1 应用外标工作曲线计算引入的不确定度u(c)
3.1.1 测量不同浓度的响应值A的随机变化引入的预估值cpred的不确定度u(cpred,A)
通常通过对若干对数值(c,A)的加权或未加权最小二乘法回归来确定工作曲线A=ac+b中的常数a和b。可利用工作曲线通过样品中被分析物产生的峰面积响应值Aobs计算出该分析样品浓度cpred。
由于A的随机变化引入的预估值cpred的不确定度可从工作曲线数据获得,适用公式(3)[8]:


式中:var(cpred)——未加权数据预估值cpred的方差;
s——残差标准偏差;
a——工作曲线斜率;
p——预估值点的进样次数,p=2;
n——工作曲线上各点测量次数之和,n=15;
b——工作曲线截距。
本实验建立的外标法工作曲线方程为A=89 794 350c-3 337.032,预估值cpred=0.003 93 mg/mL,依据式(3),(4),测量不同浓度的响应值A随机变化引入预估值cpred的不确定度u(cpred,A) =3.05×10-5mg/mL,相对标准不确定度urel(cpred,A) =u(cpred,A)/cpred=7.77×10-3。
3.1.2 工作曲线中不同浓度的标准使用液赋值引入的不确定度u(cpred,c)
u(cpred,c)来源于标准溶液的制备过程及从标准溶液到标准工作液的稀释过程。
(1)标准溶液制备引入的不确定度
根据制造商说明,本实验使用的四氢大麻酚标准品浓度c0=(100±0.5)μg/mL,服从矩形分布,标准不确定度u(c0)=0.5/=0.289(μg/mL),相对标准不确定度urel(c0)=u(c0)/c0=0.002 89。
(2)移取标准溶液引入的不确定度
本实验中,除0.01 mg/mL浓度点使用注射器从标准样品中取样并稀释,其余4种浓度的稀释均使用微量进样针从0.01 mg/mL的标准工作液中取样。以c2=0.005 mg/mL浓度点为例,这一步骤引入的不确定度来自以下三个方面:微量进样针最大容量允差引入的不确定度;微量进样针排出溶液体积引入的不确定度;温度变化引入的不确定度。
以使用250 μL进样针移取溶液为例:
本实验所用标称容量为250 μL的微量进样针已检定合格,容量允差为±1.5%[9]。按照矩形分布,不确定度分量u250,1=1.5%×250/=2.165(μL)。
操作者将吸取的溶液排出微量进样针这一过程中,排出液体积的不确定度可通过多次测量的重复性实验来评估。本实验采用移取10次称量得出的标准偏差作为不确定度计算,u250,2=0.201 μL。
容量瓶在20℃校准,实验室的温度在(20±4)℃之间变动。该影响引起的不确定度可通过计算该温度范围和体积膨胀系数来进行评估。由于液体的体积膨胀明显大于容量瓶的体积膨胀,因此只需考虑前者即可。有机液体膨胀系数1.0×10-3/℃。因此,标称容量为V针的微量进样针产生的体积变化为±V针×4×1.0×10-3。按照矩形分布,u250,3=250
将3个不确定度分量合成,使用250 μL微量进样针移取标准溶液引入的标准不确定度为=2.250 μL,相对合成标准不确定度u250,rel=u250/250=0.009 00。同理,可计算得到u100,rel=0.011 8,u50,rel=0.017 5。
(3)容量瓶定容引入的不确定度
使用容量瓶定容引入的不确定度来源于:
①校准不确定度。检定合格的1 mL A类单标线容量瓶容量允差为±0.007 mL,10 mL A类单标线容量瓶容量允差为±0.020 mL[10]。服从矩形分布,校准不确定度由最大容量允差除以求得。
②充满容量瓶至刻度的变化引起的不确定度。可通过充满该容量瓶的重复性实验来评估,并直接作为不确定度进行计算。10 mL A类单标线容量瓶的重复性不确定度为0.006 41 mL,1 mL A类单标线容量瓶的重复性不确定度为0.001 62 mL。
③温度引起体积变化引入的不确定度。计算方法同3.1.2(2)。
将三个不确定度分量合成,计算得10 mL容量瓶定容引入的相对标准不确定度u10,rel=0.002 66,同理,可计算得1 mL容量瓶定容引入的相对标准不确定度u1,rel=0.004 93。
(4)引起标准浓度赋值ci误差随机效应的合成
由标准值不确定度引起的预估值cpred的不确定度u(cpred,ci)近似值为u(ci)/n,n是用于校准的ci值的数目(本实验n=5)[8]。
对于c1=0.01 mg/mL的浓度点,合成标准不确定度=3.93×10-5mg/mL,u(cpred,c1)=uc(c1)/n =7.86×10-6mg/mL。
c2,c3,c4,c5各浓度点的标准值ci=c1×吸取体积/定容体积。以c2=0.005 mg/mL浓度点为例,其相对合成标准不确定度uc,rel(c2)=0.014 2,合成标准不确定度uc(c2)=uc,rel(c2)×c2=7.10×10-5mg/mL,u(cpred,c2) =uc(c2)/n =1.42×10-5mg/mL。依此计算,得u(cpred,c3),u(cpred,c4),u(cpred,c5)分别为7.14×10-6, 2.68×10-6,1.86×10-6mg/mL,则导致标准浓度赋值ci误差的合成标准不确定度u(cpred,c)按式(5)计算:
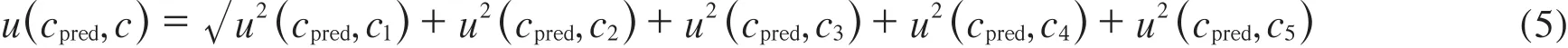
将相关数据代入式(5)得u(cpred,c)=1.80×10-5mg/mL。
3.1.3 应用工作曲线计算引入的不确定度合成
合成3.1.1与3.1.2节计算得到的两个分量,得到用工作曲线计算引入的不确定度u(cpred)

3.2 称量样品引入的不确定度u(m)
本实验所用天平的计量证书标明其称量的最大允差为0.09 mg,称量重复性(变动性)为0.04 mg(依据天平校准证书),包含因子k=,标准不确定度。净重由两次称量操作所得(一次为皮重,另一次为毛重),每一次称量均为独立观测结果,合成不确定度
3.3 样品稀释过程引入的不确定度
溶解样品加入溶剂甲醇的体积V1、从甲醇一次提取液中移取V2体积的样品溶液、再次稀释加入的溶剂体积V3这三项引入的不确定度均源于移液器最大容量允许误差、移液器排出体积的不确定性以及温度变化这3个方面。合成标准不确定度计算方法见3.1.2(2),计算结果见表1。
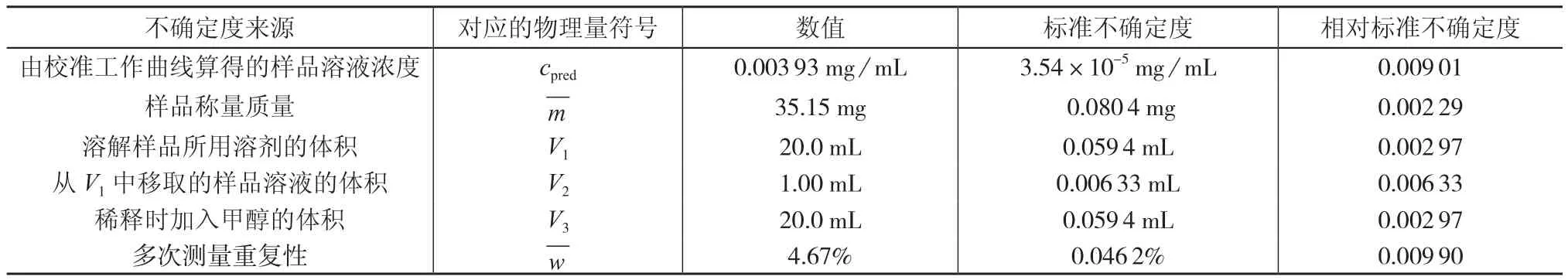
表1 各不确定度分量表
3.4 测量重复性引入的不确定度urep
在重复性条件下,对同一样品从取样开始重复测量n次,n次测定的平均值作为结果,则测量重复性引入的不确定度,相对标准不确定度,计算结果见表1。
4 合成标准不确定度的计算
将表1中各不确定度分量按式(6)合成,求得w的相对标准不确定度uc,rel(w):
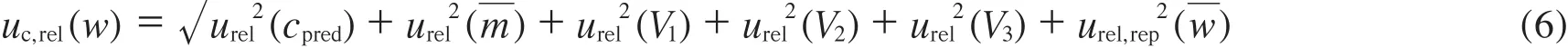

5 扩展不确定度的计算及测量结果表示
95%置信概率下,取包含因子k=2,扩展不确定度U=k×uc(w)%=0.15%,样品中四氢大麻酚的含量表示为(4.67±0.15)%,k=2。
6 结语
确定预估值cpred不确定度的标准方法有三种[8]:从计算所得的方差和协方差来获取;从校准数据获取;从用来导出工作曲线的软件提供的信息获取。第一种方法计算繁琐,第三种不具有普适性,而第二种方法不但方法相对简便可行,公式易于理解,而且通过引入残差标准偏差间接反映出线性相关系数的大小,使得不确定度的评定更加可靠,因此笔者认为第二种方法为首选方法。
实验结果表明,采用高效液相色谱外标工作曲线法测定大麻树脂中四氢大麻酚含量,测量结果的不确定度主要是由外标工作曲线以及多份样品的重复测量引起的,重复性误差对小体积移液器或容量瓶影响较大,稀释样品步骤直接造成不确定度增大,采用较大体积的移液器和容量瓶可减小不确定度。
[1] JJF 1059-1999 测量不确定度评定与表示[S].
[2] 王威,倪春芳,张润生.高效液相色谱法测定海洛因含量的不确定度评定[J].中国司法鉴定,2008(2): 28-30.
[3] 屈颖. 布洛芬缓释胶囊中布洛芬含量的测量不确定度评估[J].天津药学,2008,20(1): 8-10.
[4] 汪辉,曹小彦,彭新凯,等.高效液相色谱法测定小麦粉与大米粉中甲醛次硫酸氢钠含量的不确定度评定[J].食品科学,2009,30(12): 205-208.
[5] 李东刚,李春娟,吴凡,等.高效液相色谱法测定巴氏杀菌乳中糠氨酸不确定度的评定[J].化学分析计量,2008,17(3): 7-9.
[6] 徐靖,汪欢晃,刘娟,等.超高效液相色谱法测定化妆品中孕激素的不确定度[J].化学分析计量,2008,18(3): 11-14.
[7] 李志梅,孙东卫.外标法测定饮用水中无机元素的[J].中国计量,2009(8): 72-73.
[8] CNAS-GL06 化学分析中不确定度的评估指南[S].
[9] JJG 196-2006 常用玻璃量器检定规程[S].
[10] JJG 646-2006 移液器检定规程[S].
Uncertainty Evaluation of Determination of Tetrahydrocannabinol in Cannabis Resin by HPLC External Standard Working Curve Method
Zhai Wanfeng
(Department of Forensic Science, Chinese People’s Public Security University, Beijing 100038, China)
Zhang Chunshui, Gao Lisheng
(Institute of Forensic Science Ministry of Public Security P.R.C, Beijing 100038, China)
The determination uncertainty of tetrahydrocannabinol in cannabis resin by HPLC external standard working curve method was evaluated based on JJF 1059-1999 Evaluation and Expression of Uncertainty in Measurement. The mathematical models to evaluate the uncertainty of quantitative results of HPLC external standard working curve method were established, and each source and uncertainty component was analysized, and the combined expanded uncertainty was 0.15%(k=2).
HPLC; external standard method; uncertainty; cannabis resin
O657.7
:A
:1008-6145(2012)05-0008-04
10.3969/j.issn.1008-6145.2012.05.002
*公安部重点研究计划支持项目(2009ZDYJGAES024);中国人民公安大学学科建设与研究生创新项目(YX11120)
联系人:翟晚枫;E-mail: betty871211.student@sina.com
2012–07–22
