铁(氢)氧化物悬液中磷酸盐的吸附-解吸特性研究
2012-06-26王小明孙世发谭文峰胡红青冯雄汉
王小明, 孙世发, 刘 凡, 谭文峰, 胡红青, 冯雄汉
铁(氢)氧化物悬液中磷酸盐的吸附-解吸特性研究
王小明, 孙世发, 刘 凡, 谭文峰, 胡红青, 冯雄汉*
(农业部亚热带农业资源与环境重点实验室, 华中农业大学 资源与环境学院, 湖北 武汉 430070)
铁(氢)氧化物对P的吸持和释放在一定程度上决定着P的生物有效性和水体富营养化。以两种环境中常见晶质铁氧化物(针铁矿和赤铁矿)为对照, 采用X射线衍射(XRD)、透射电镜(TEM)、热重分析(TGA)和孔径分析以及动力学和吸附-解吸热力学平衡等技术方法, 研究了弱晶质水铁矿对P吸附-解吸特性, 并探讨了相关机制。实验表明, 三种矿物对P的吸附分为起始的快速反应和随后的慢速反应, 它们均符合准一级动力学过程, 反应中OH–释放明显滞后于P吸附, P吸附经历了从外圈到内圈配位、单齿到多齿配位过渡的过程; 与晶质氧化铁比,水铁矿吸附容量和OH–释放量更大、慢速吸附反应更快、存在缓慢扩散反应阶段, 吸附容量依次是: 水铁矿(4.36 μmol/m2) >> 针铁矿(2.62 μmol/m2)>赤铁矿(2.28 μmol/m2); 针铁矿和赤铁矿吸附P符合L (Langmuir)模型, 而水铁矿更符合F (Freundlich)模型。中性盐介质(KCl)中在最大吸附量时P的解吸率依次为: 水铁矿(8.5%) < 针铁矿(10%) < 赤铁矿(12.5%); 柠檬酸通过配体解吸和诱导溶解两种机制促进P的解吸, 最大吸附量时解吸率依次是: 针铁矿(25%) < 水铁矿(32%) < 赤铁矿(50%)。
铁(氢)氧化物; 水铁矿; 针铁矿; 赤铁矿; 磷; 吸附和解吸
0 引 言
P是植物生长的必需营养元素, 也是造成富营养化的污染元素, 主要来源于土壤和沉积物。我国缺P土壤面积约占耕地的一半,但多数土壤中全P含量并不低[1]。植物缺P一般有两方面原因: 土壤P含量和有效性低; 施入的P被土壤吸持, P利用率很低[2]。因此, 如何提高P的生物有效性, 同时避免P流失造成环境污染备受关注。
铁氧化物(包括氧化物、氢氧化物和水合氧化物)表面OH含量高, 环境pH下表面带可变正电荷, 故可以通过内圈配位(OH配位交换)和外圈配位(静电作用)吸附P, 在较大程度上影响P的形态、迁移和转化[3]。土壤P的有效性与铁、铝氧化物密切相关, 土壤难释放P主要有铁、铝结合态磷(Al-P、Fe-P)、铁铝氧化物包裹的闭蓄态磷(O-P)和钙结合态磷(Ca-P), 南方土壤以前两者为主[4]。氧化铁对P的吸附受pH、离子强度和共存离子等因素的影响。吸附量随pH升高而逐渐下降[5]。水铁矿在pH 4~7.5对P的吸附与离子强度无关, 这可能与内圈表面配位有关[6–7]。此外, 环境中的胡敏酸(HA)、富里酸(FA)和柠檬酸等有机酸对铁氧化物吸附P有明显的抑制作用, 这与形成空间位阻和竞争表面吸附位点有 关[8–9]。铁氧化物吸附P在初始阶段很迅速, 随后紧接着会进入一个缓慢阶段, 直到吸附解吸平衡, 弱晶态铁氧化物的缓慢扩散吸附反应更明显且存在部分不可逆吸附位点[10–11]。植物在缺P时可分泌柠檬酸、草酸等有机酸, 它们可通过配位交换和诱导配位体溶解促进铁氧化物释放P[12]。
通过光谱学技术对晶质氧化铁吸附P的机制已有深入研究。铁氧化物吸附P以质子化((FeO)2(OH)PO)和非质子化((FeO)2PO2)的双齿双核表面配位为主[13–14]。一定pH和P吸附量条件下, 还存在少量非质子化的单齿表面配位[15–16]。然而, 氧化铁的结构类型对其吸附P的差异和机制探讨却较少。尤其是针对环境中广泛分布的弱晶质氧化铁与P的表面作用特性和机制仍鲜见报道。而土壤中氧化铁类型差异是引起P有效性显著不同的主要原因[17–18]。水铁矿是环境中最为常见的弱晶质铁氧化物, 是晶质铁氧化物形成的前驱物, 比表面积大、表面活性高, 对环境中污染物质和营养元素的地球化学循环过程起着重要作用[19–21]。文研究以针铁矿和赤铁矿为参照, 对比研究水铁矿吸附P的动力学过程以及吸附和解吸P的特异性, 结合矿物的结构特点、表面位点、热重和孔径特点等分析探讨相关机制,为不同类型土壤中如何调控P的有效性及理解P元素的地球化学循环提供科学依据。
1 材料与方法
1.1 三种不同铁氧化物的合成[22]
水铁矿 搅拌条件下, 将40 g Fe(NO3)3·9H2O溶于500 mL去离子水中, 逐滴加入1 mol/L NaOH约330 mL, 调节pH 7~8。洗涤, 离心, 冻干, 磨碎, 过60目筛, 于4 ℃冰箱中储存。
针铁矿 搅拌条件下, 将180 mL 5 mol/L NaOH溶液加到100 mL 1 mol/L Fe(NO3)3溶液中, 用去离子水稀释至2 L, 密闭, 70 ℃老化60 h。洗涤, 离心, 烘干, 磨碎, 过60 目筛, 于干燥器中备用。
赤铁矿 搅拌条件下, 将40 g Fe(NO3)3·9H2O溶于500 mL预加热至90 ℃的去离子水中,分别加入预加热至90 ℃的1 mol/L KOH 溶液300 mL和 1 mol/L NaHCO3溶液50 mL, 密闭, 90 ℃老化48 h。洗涤, 离心, 烘干, 磨碎, 过60目筛, 于干燥器中备用。
1.2 样品分析与鉴定
粉晶X射线衍射 矿物按粉末压片法在Bruker D8 Advance上进行XRD分析。测试条件为: Cu辐射, 管压40 kV, 管流40 mA, 扫描速度4˚/min, 步长0.02。
透射电子显微镜 在无水乙醇中加入少量样品, 超声分散, 用微型吸管吸取少量悬液滴到已镀碳的Cu网上, 室温晾干, 用H-7650透射电子显微镜分析。测试条件: 加速电压100 kV, 线分辨率0.204 nm。
热重分析 在NETZSCH STA 449C上进行热重分析。样品质量约10 mg, N2流量为20 mL/min, 升温速率为10 ℃/min。
孔径分析 用Quantachrome Autosorb-1进行比表面和孔径分布分析。水铁矿在50 ℃脱气12 h, 针铁矿和赤铁矿在80 ℃脱气12 h, 去除水和其他吸附质。然后在77 K、相对压力(/0)分别为10–7~0.9962和0.9962~0.0497范围内进行N2等温吸附/脱附实验。比表面积根据多点Brunauer-Emmett-Teller(BET)方法计算, 用t方法(选取相对压力为0.4~0.6)计算样品的微孔体积、微孔面积和外表面积。根据N2等温吸附曲线, 用Saito-Foley(SF)方法分析矿物的孔径分布, 根据接近饱和压力(/0≈ 0.98)时的N2吸附量估算总孔体积。
1.3 P吸附动力学实验
水铁矿、针铁矿和赤铁矿分别配成0.36 g/L、2.8 g/L和4 g/L悬液, 用自动电位滴定仪(瑞士万通907智能型)维持pH 4.5, 平衡16 h。配制浓度为300 mg/L KH2PO4, 调节pH 4.5。将20 mL P溶液与悬液混合, 使得体系初始P浓度为50 mg/L, 用滴定仪STAT模式加酸不断维持pH 4.5, 加酸的摩尔量即为反应过程中OH–释放量。不同时间点取样, 每次2.5 mL, 过0.22 μm微孔滤膜过滤, P浓度用钼锑抗分光光度法测定。
1.4 P吸附与解吸实验
水铁矿、针铁矿和赤铁矿分别配成2 g/L、20 g/L和20 g/L悬液, 用0.1 mol/L HCl和 KOH调节pH 4.5,平衡16 h。配制1 g/L KH2PO4和0.01 mol/L KCl, 调节pH 4.5。然后往50 mL塑料离心管(记录空管质量)中分别加水铁矿、针铁矿和赤铁矿悬液4.525 mL、 3.53 mL和5 mL(控制比表面积一致), 再分别加0~7.5 mL 1 g/L KH2PO4, 用0.01 mol/L KCl定容至30 mL,使得P浓度在0~250 mg/L变化。25 ℃和200 r/min条件下振荡24 h, 于0.5 h、16 h和24 h将pH调至4.5,记录加酸量, 加酸的总量即为吸附过程中OH–释放量。反应结束后离心, 取上清液经0.22 μm滤膜过滤。实验做2组平行, 取平均值。吸附结束, 倾出上清液, 称量管重, 计算残留液质量。随后, 向平行1中加入10 mL 0.01 mol/L柠檬酸溶液, 用蒸馏水定容至30 mL; 平行2直接用0.01 mol/L KCl定容至30 mL。摇匀, 调节pH 4.5, 振荡14 h, 解吸后pH调回4.5, 离心, 取清液经0.22 μm滤膜过滤。P和Fe浓度分别用钼锑抗和邻二氮菲分光光度法测定。
2 结果与讨论
2.1 矿物的鉴定和表征
图1是三种矿物的XRD图谱, 水铁矿的主要特征峰晶面间距和对应的衍射面网(): 0.250 nm (110)和0.148 nm(300),为弱晶质二线型水铁矿; 针铁矿的为: 0.498 nm(020), 0.418(110), 0.338(120), 0.269(130), 0.258(021), 0.245(111), 0.225(121), 0.219(140), 0.172(221)和0.156(151)等; 赤铁矿的为: 0.367 nm(012), 0.269(104), 0.251(110), 0.220(113), 0.184(024), 0.169(116), 0.159(018), 0.148(214)和0.145(300)。它们的衍射数据与标准衍射卡片相吻合,无其他衍射峰,卡片编号分别是00-029-0712(水铁矿)、01-081-0464(针铁矿)和01-089-8104(赤铁矿)。故合成的样品分别为水铁矿、针铁矿和赤铁矿单相氧化铁。
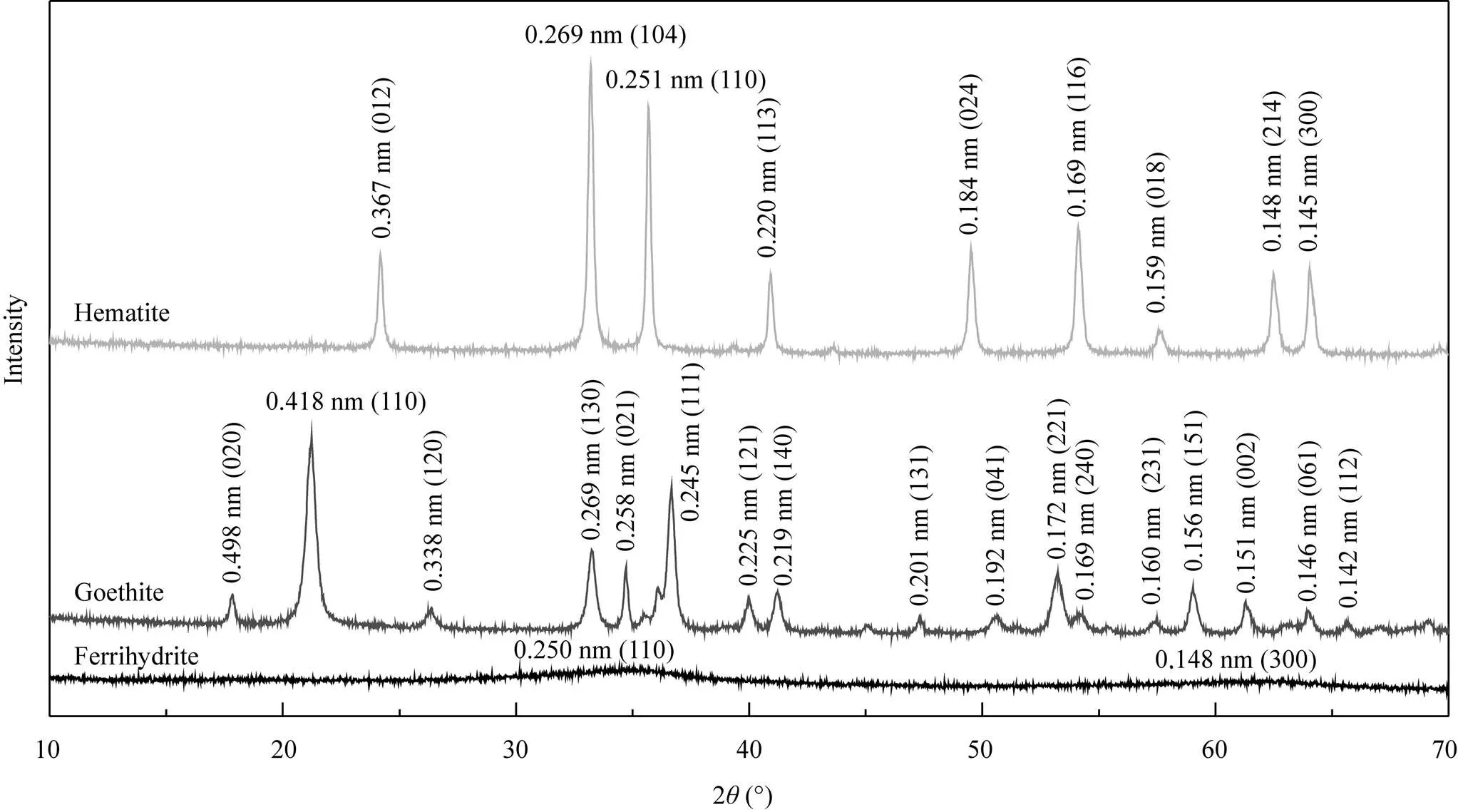
图1 三种矿物的X射线衍射(XRD)图谱
三种矿物的透射电子显微镜图片见图2。可以看出, 三种矿物具有均一的特征形貌, 进一步证实上述样品均为单相矿物。水铁矿结晶弱, 晶粒小, 以无定形的球形颗粒聚合体形式存在, 单个球形水铁矿颗粒尺寸大约是2~6 nm (图2a), 与Abou-Hassan.得到的水铁矿的粒径大小相近[23]; 针铁矿的形貌是针状, 结晶长度为1 μm左右, 宽度为40 nm左右(图2b); 赤铁矿为菱形粒状晶体, 分散性较好, 结晶尺寸为50 nm左右(图2c)。

图2 水铁矿(a)、针铁矿(b)和赤铁矿(c)的透射电子显微镜(TEM)图片
图3是三种矿物的热重及相应的微分曲线。水铁矿在约92 ℃有明显的失重峰, 在256 ℃和356 ℃有微弱的失重峰, 总失重约21.2%; 针铁矿在250 ℃有非常明显的失重峰, 失重约25%; 赤铁矿没有明显的失重峰, 失重仅5.5%。铁氧化物的失重一般由吸附水(100 ℃左右)、结构OH(200~600 ℃)及其他一些吸附物质丢失引起。水铁矿吸附水(30~150 ℃)和OH(150~450 ℃)比例分别占11%和7.7%, 表明水铁矿中含有大量吸附水和一定量OH。结构中水分子分布于纳米孔状结构, 通过H键与周围阴离子连接[24], 或通过微弱的物理化学作用力吸附在水铁矿表面, 且这些水分子连接着小颗粒使水铁矿常以聚集体形式存在[25]。针铁矿吸附水和OH比例分别占1.4% (30~150 ℃)和13.5% (150~350 ℃), 表明针铁矿中含大量OH和极少量吸附水。赤铁矿失重比例较小, 意味着仅含少量的H2O和OH。这些矿物的不同结构特点(OH位点密度)预示着它们的吸附容量和反应活性会有明显不同。

图3 三种矿物的热重分析(TGA)及微分曲线图
三种矿物的孔性结构参数见表1。它们的总孔体积依次是水铁矿(0.2739 cm3/g) > 赤铁矿(0.2317 cm3/g) > 针铁矿(0.1987 cm3/g),平均孔径依次是水铁矿(29.13Å) < 针铁矿(169.7Å) < 针铁矿(279.2Å), 表明水铁矿孔隙发达且多以微孔形式存在, 而针铁矿和赤铁矿的孔隙较少且多以大孔形式存在。水铁矿的微孔体积和微孔面积分别是0.1628 cm3/g和249.8 m2/g, 结晶度良好且形貌规则的针铁矿和赤铁矿则几乎不存在微孔。多点BET法测得的三种矿物的比表面积依次是水铁矿(347.6 m2/g) > 针铁矿(44.58 m2/g) > 针铁矿(31.45 m2/g)。水铁矿大比表面积与它的小尺寸和弱结晶特性有关, 利用BET-N2方法获得的常见值一般为200~350 m2/g[26]。
2.2 三种矿物吸附P的动力学特性
三种矿物吸附P(图4a)和吸附过程中OH–释放(图4b)的动力学曲线如图所示。P吸附和OH–释放过程都表现出初始的快速反应和紧接的慢速反应, 它们均符合一级反应动力学过程(图5)。环境中矿物吸附污染物质或营养元素一般有四个阶段: 初始快速吸附反应、慢速吸附反应、持续缓慢扩散反应和吸附解吸平衡反应[27]。前20 min属于快速吸附反应阶段, 吸附反应速率相当, 速率常数分别是: 3.13E-3(水铁矿)、3.08E-3(针铁矿)和3.05E-3(赤铁矿); 吸附量分别2.03 μmol/m2(水铁矿)、1.93 μmol/m2(针铁矿)和1.47 μmol/m2(针铁矿); 三种矿物紧接的慢速反应差异较大, 水铁矿和针铁矿的慢速反应发生在20~240 min, 而赤铁矿持续到720 min,速率常数依次是: 水铁矿(2.60E-4) > 针铁矿(1.07E-4) > 赤铁矿(4.01E-5), 吸附量分别是2.96 μmol/m2(水铁矿)、2.26 μmol/m2(针铁矿)和1.78 μmol/m2(赤铁矿)。此外, 水铁矿在240~1800 min时间里表现出持续的缓慢扩散反应, 速率常数2.82E-5。缓慢扩散反应受矿物结晶度和颗粒形貌影响[28], 水铁矿结晶弱, 微孔体积和微孔面积大(表1),故缓慢扩散反应明显, 而针铁矿和赤铁矿因结晶度高, 形貌规则和微孔少, 没有出现缓慢扩散反应(图5b); 1800 min时吸附量分别是: 3.6 μmol/m2(水铁矿)、2.36 μmol/m2(针铁矿)和2.01 μmol/m2(针铁矿)。因此, 相比针铁矿和赤铁矿, 水铁矿有更大的吸附容量、OH–释放量和更快的吸附反应速率且存在持续缓慢扩散吸附反应阶段, 这应与水铁矿较高OH–位点密度、弱结晶性和微孔扩散密切相关。
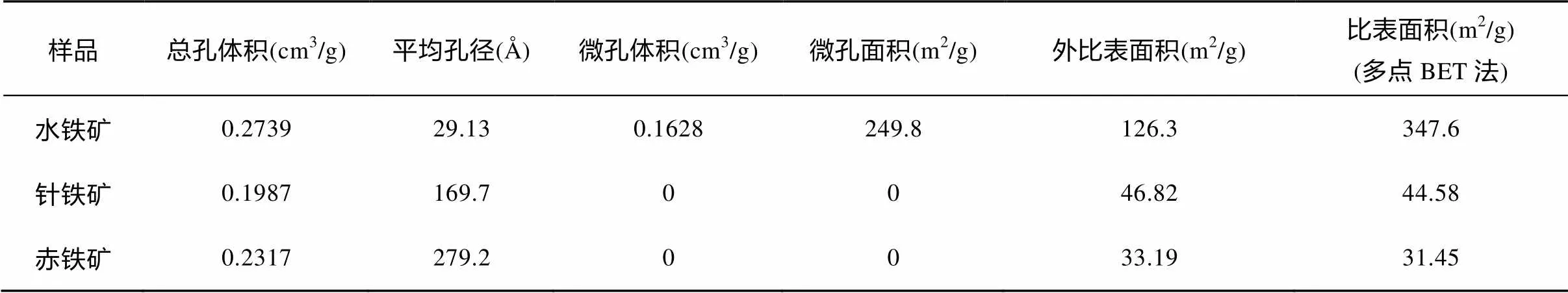
表1 三种矿物孔性结构参数
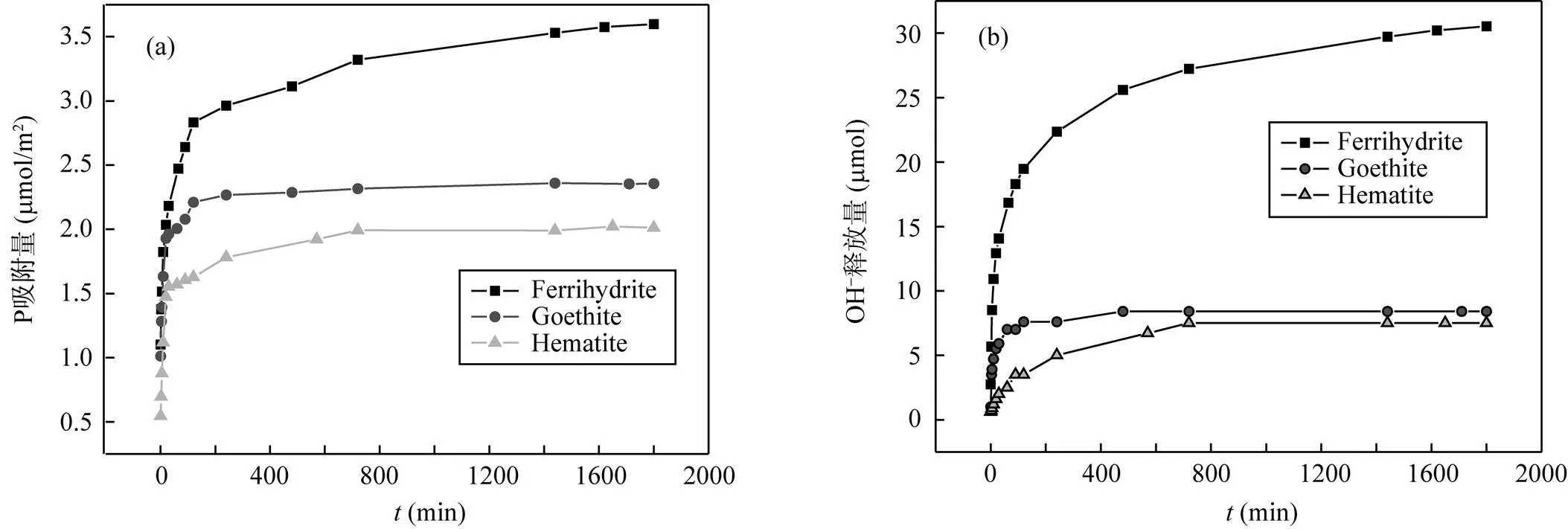
图4 三种矿物吸附P (a)及吸附P过程OH–释放(b)动力学曲线
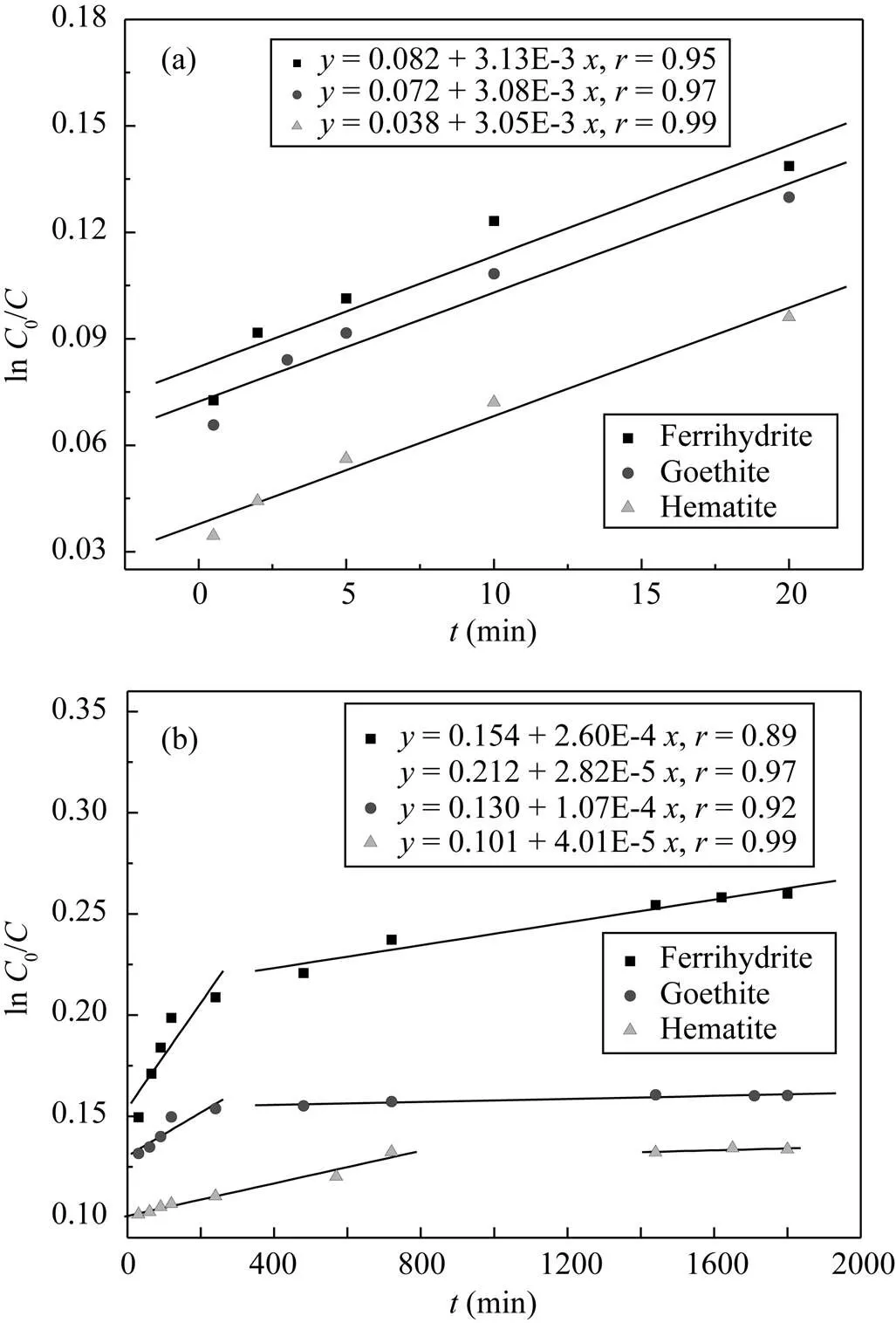
图5 三种矿物在初始20 min (a)和随后30~1800 min (b)吸附P的准一级动力学拟合
图6是三种矿物OH–释放量与P吸附量摩尔比(OH–/P)随吸附量的变化曲线。OH–/P先随吸附量的增加而急剧增加, 然后逐渐趋于平衡, 此时OH–释放与P吸附接近同步增加; 随吸附量的继续增大, 针铁矿和赤铁矿将该平衡的OH–/P保持至饱和吸附(比例约0.3), 而水铁矿又经历一个缓慢的OH–/P上升阶段后才趋于平衡(比例约0.7), 表明铁氧化物吸附P过程中OH–释放明显滞后于P吸附, 且随吸附反应的进行OH–/P逐渐增加, 这意味着铁氧化物吸附P动力学过程是: 先通过外圈配合物吸附P, 然后由外圈配合物逐渐向内圈配合物转化,转化过程先形成单齿配合物, 后形成双齿配合物, 前人也描述过相近的动力学过程[29]。对于水铁矿的OH–/P后一缓慢增加阶段的原因, 可归结为水铁矿是一类弱晶质体, 表面位点不均一, P与聚合体内部位点结合后OH–没能及时释放。Willett.通过电子微探针证实P可迁移进入水铁矿聚合体内部位点[30]。
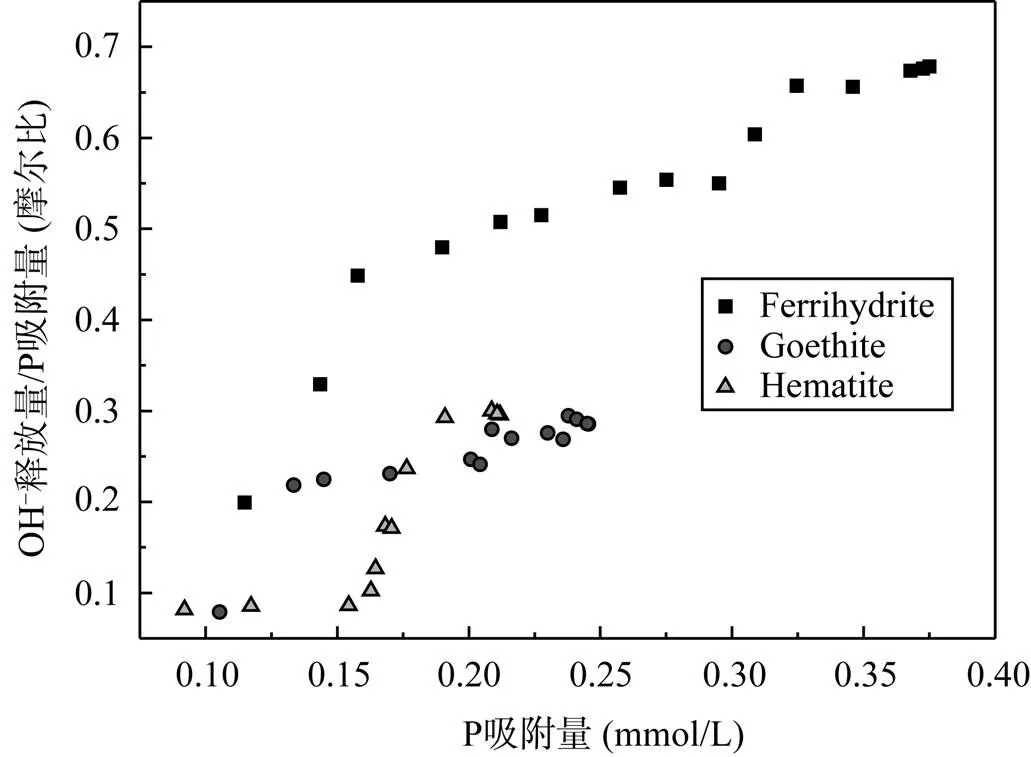
图6 三种矿物的OH–释放量与P吸附量的摩尔比(OH–/P)随吸附量变化曲线
2.3 三种矿物吸附P等温线
三种矿物吸附P的等温吸附曲线见图7。无论单位比表面积(图7a)还是单位质量(图7b), 水铁矿的吸附容量都远高于针铁矿和赤铁矿, 且动力学数据表明, 弱晶质水铁矿在24 h内未完全达到吸附饱和, 故水铁矿的实际平衡吸附量会更高。通过Langmuir拟合, 水铁矿、针铁矿和赤铁矿最大吸附容量依次是: 4.36 μmol/m2(1514 μmol/g) >> 2.62 μmol/m2(117 μmol/g) > 2.28 μmol/m2(71 μmol/g), 这与报道的相近(水铁矿4.54 μmol/m2 [31], 针铁矿2.6 μmol/m2 [17])。此外, 与针铁矿和赤铁矿相比, 水铁矿的吸附等温线更符合F型, 也与其吸附P表现出的动力学特点相符, 这应与其表面具有更高的不均一性有关[32]。
水铁矿高的吸附容量与它位点密度和表面结构相关。热重结果表明水铁矿中含有大量吸附水, 在溶液状态时, 矿物中水分子很容易羟基化解离, 因而具有较高的单配位OH位点密度(6.00 nm-2)[33]; 针铁矿的单配位OH位点密度是3.125 nm-2 [34], 故吸附容量与位点密度有正相关关系。Zhao.[25]研究表明, 水铁矿结构中有25%四面体配位Fe, 且分布在颗粒表面, 主体结构仍是八面体配位Fe。颗粒表面的四面体位点是一种不饱和配位位点, 暴露在空气中容易吸附水而增加平均配位数。因此, 外来离子不仅可以占据不饱和配位位点(CUS), 替代水铁矿中水解离后的OH位点, 也可以通过微孔扩散与内部隐藏位点结合, 从而表现出更大的吸附容量和OH–释放量。据报道, 天然和合成的针铁矿有相对恒定的吸附容量, 主要是在110面形成双核配位, 最大吸附容量大约是2.51 μmol/m2 [35]。赤铁矿的吸附容量(0.19~3.33 μmol/m2)与形貌和结晶度密切相 关[36]。
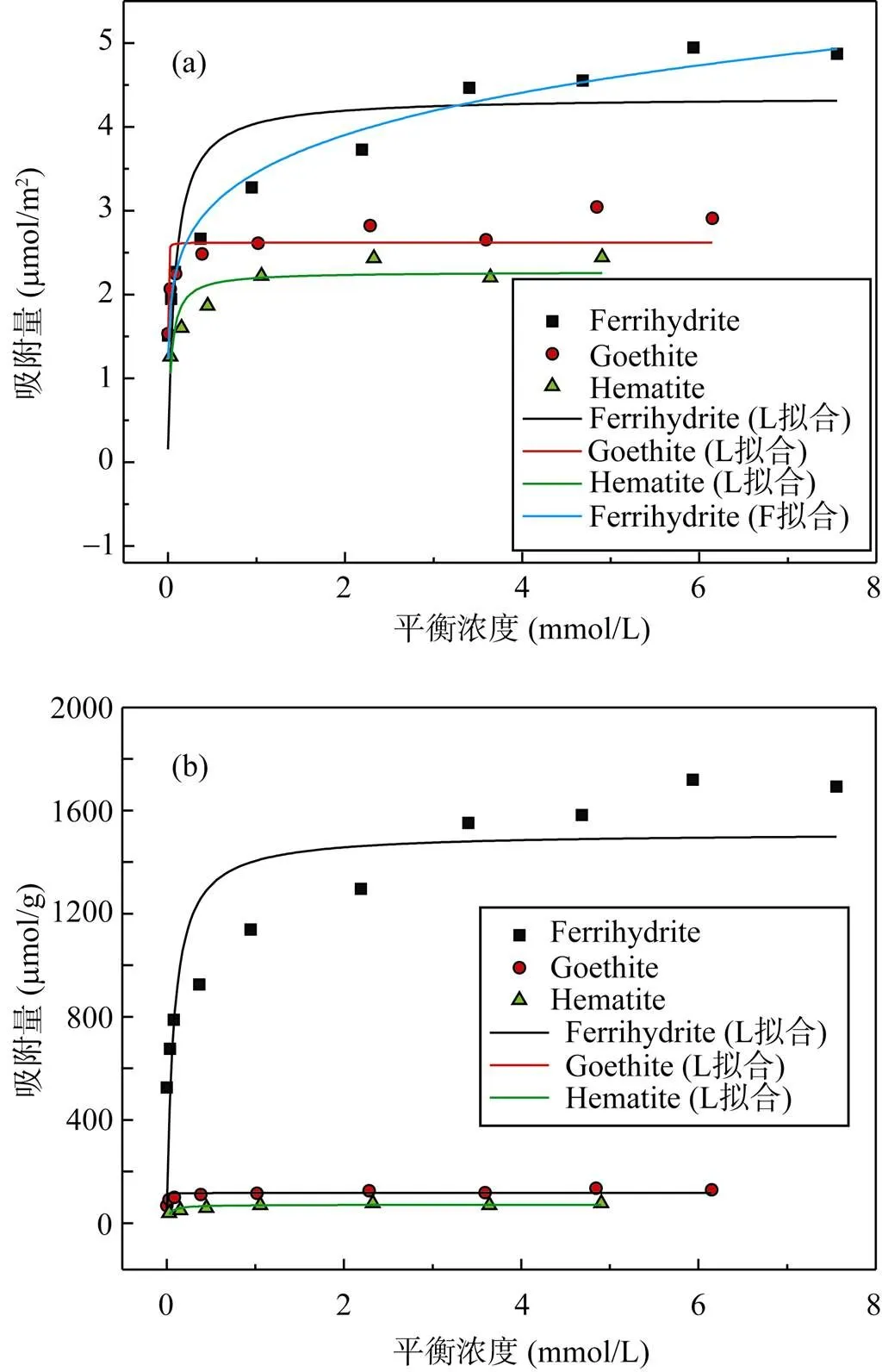
图7 三种矿物单位比表面积(a)和单位质量(b)Langmuir或Freundlich拟合吸附等温线
2.4 三种矿物表面吸附P的解吸
图8和图9分别是三种矿物吸附P后在KCl和柠檬酸介质中解吸量和解吸率随吸附量的变化曲线。三种矿物的P解吸量与吸附量之间有很好的线性关系, 直线的斜率代表了矿物解吸P的难易程度, 斜率越小越难解吸。KCl介质中斜率大小为: 0.279(赤铁矿) > 0.220(针铁矿) > 0.140(水铁矿); 柠檬酸介质中为: 0.554(赤铁矿) > 0.297(针铁矿) > 0.233(水铁矿) (图8和图9a)。KCl中三种矿物表面吸附P的解吸率都较低,随P吸附量的增加先快速增加, 后趋于平衡(图8b)。若将KCl解吸的P视为非专性吸附态, 表明三种矿物均以专性吸附P为主, 非专性吸附比例随吸附量增加而增大, 最大时占10 %左右。KCl介质中最大吸附量时解吸率依次是: 水铁矿(8.5%) < 针铁矿(10%) < 赤铁矿(12.5%), 这与文献报道的顺序一致[37]。KCl介质中水铁矿P解吸率最低, 表明水铁矿中专性吸附P的比例最大, 这是由其更高的的表面位点密度和表面活性决定的。在低浓度时P优先结合高活性表面位点, 以专性吸附为主, 因此解吸率较低; 随着P含量增加, 占据低活性位点的非专性吸附比例相对增加, 解吸率增加; 再继续增加P时, 表面位点吸附接近饱和, 少量P扩散进入铁氧化物微孔或介孔结构中, 使解吸率基本维持不变。
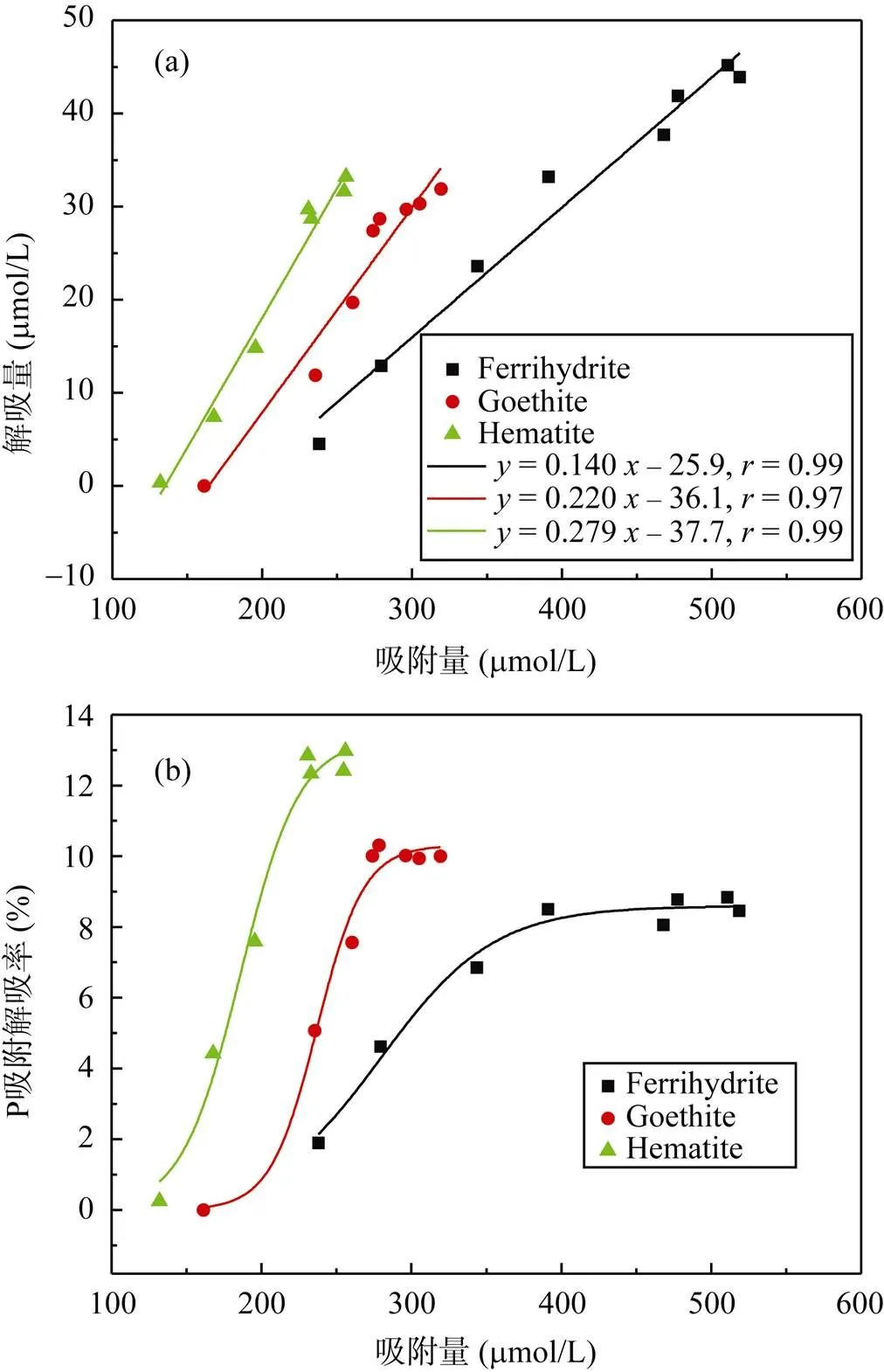
图8 在0.01 mol/L KCl中P解吸量(a)和吸附解吸率(b)随吸附量变化曲线
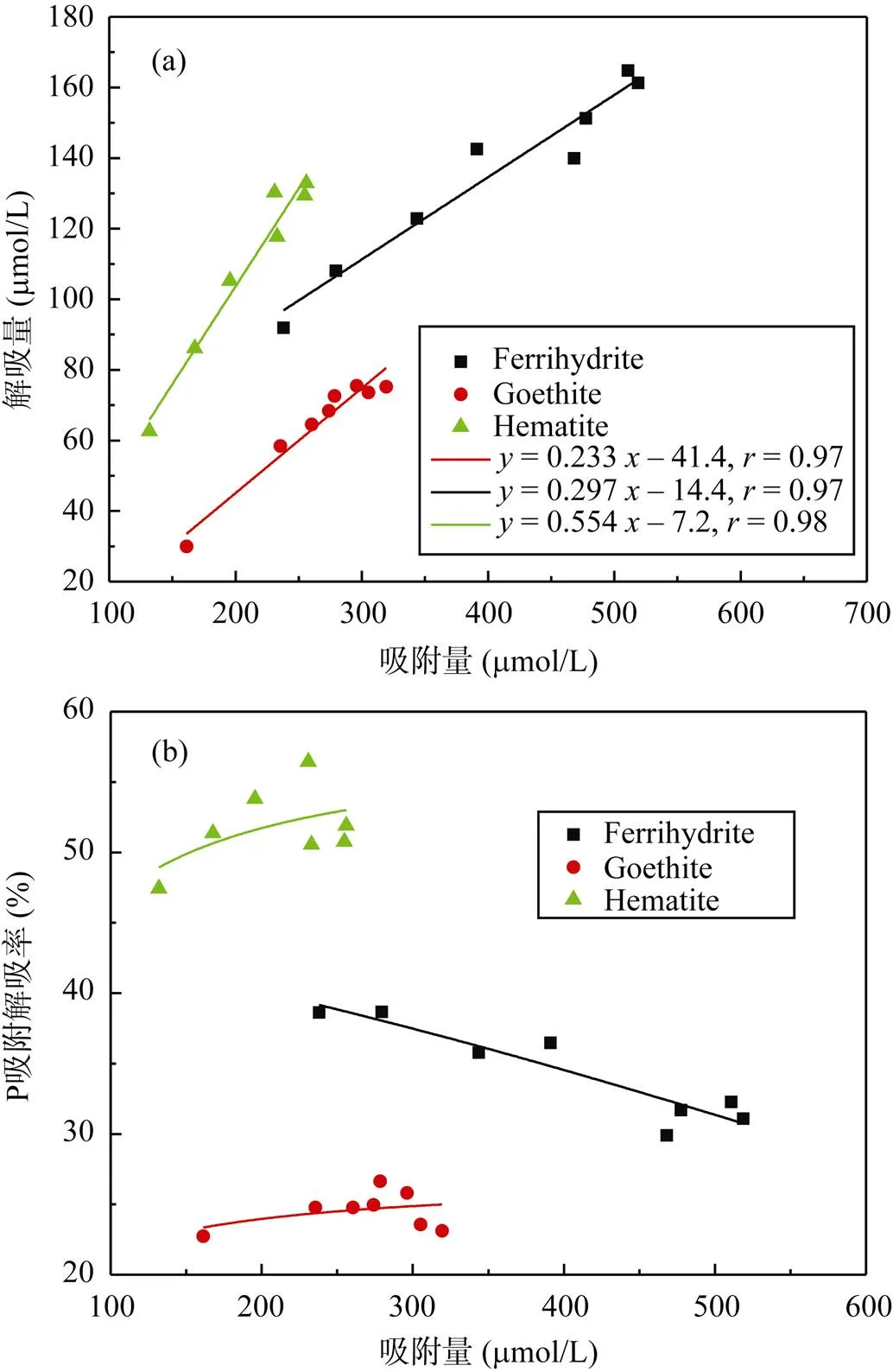
图9 在3.33 mmol/L柠檬酸中解吸量(a)和 P吸附解吸率(b)随吸附量变化曲线
柠檬酸对铁氧化物解吸P有明显的诱导效应, 使得P的解吸率较高, 且针铁矿和赤铁矿的解吸率随P吸附量增加缓慢增加, 而水铁矿缓慢降低, 这可能与水铁矿结晶弱, 部分P通过扩散进入聚合体及微孔内部难以解吸有关。最大吸附容量时解吸率依次是: 针铁矿(25%) < 水铁矿(32%) < 赤铁矿(50%) (图9b)。柠檬酸主要通过配位交换(式1)和诱导配位体溶解(式2)释放P, 配位交换是有机阴离子与矿物表面位点上的P交换, 从而释放P; 诱导配位体溶解是有机配体先吸附在矿物表面Fe结构位点, 造成矿物表面缓慢溶解, 溶解的Fe和释放的P形成铁磷络合物[12]。两种机制都包含表面铁络合物的形成, 无论是哪一种机制, 有机酸促进P的释放既与铁氧化物表面区域的结构稳定性有关, 还和表面Fe位点以及溶解的三价铁络合物的稳定性有关[38]。水铁矿、赤铁矿和针铁矿解吸液中Fe3+含量依次是 150 μmol/L >> 20 μmol/L > 10 μmol/L (图10a), 表明柠檬酸诱导水铁矿溶解比赤铁矿和针铁矿显著得多,水铁矿通过溶解释放的P远多于针铁矿和赤铁矿, 解吸率大约5% (图10b)。显著性差异分析表明, 图中柠檬酸对P解吸率显著大于KCl的(0.01 < P = 0.0283 < 0.05)。因此, 有机酸的分泌对以弱晶态铁氧化物为主的土壤中P生物有效性提高有重要意义。环境中铁氧化物作为土壤中吸持P的重要载体, 在维持土壤-沉积物-水体中P的平衡发挥着重要作用。不同氧化铁矿物对P吸附-解吸特点的差异可以为今后更好地提高土壤中P生物有效性和P的环境地球化学循环提供一些理论依据和数据支持。
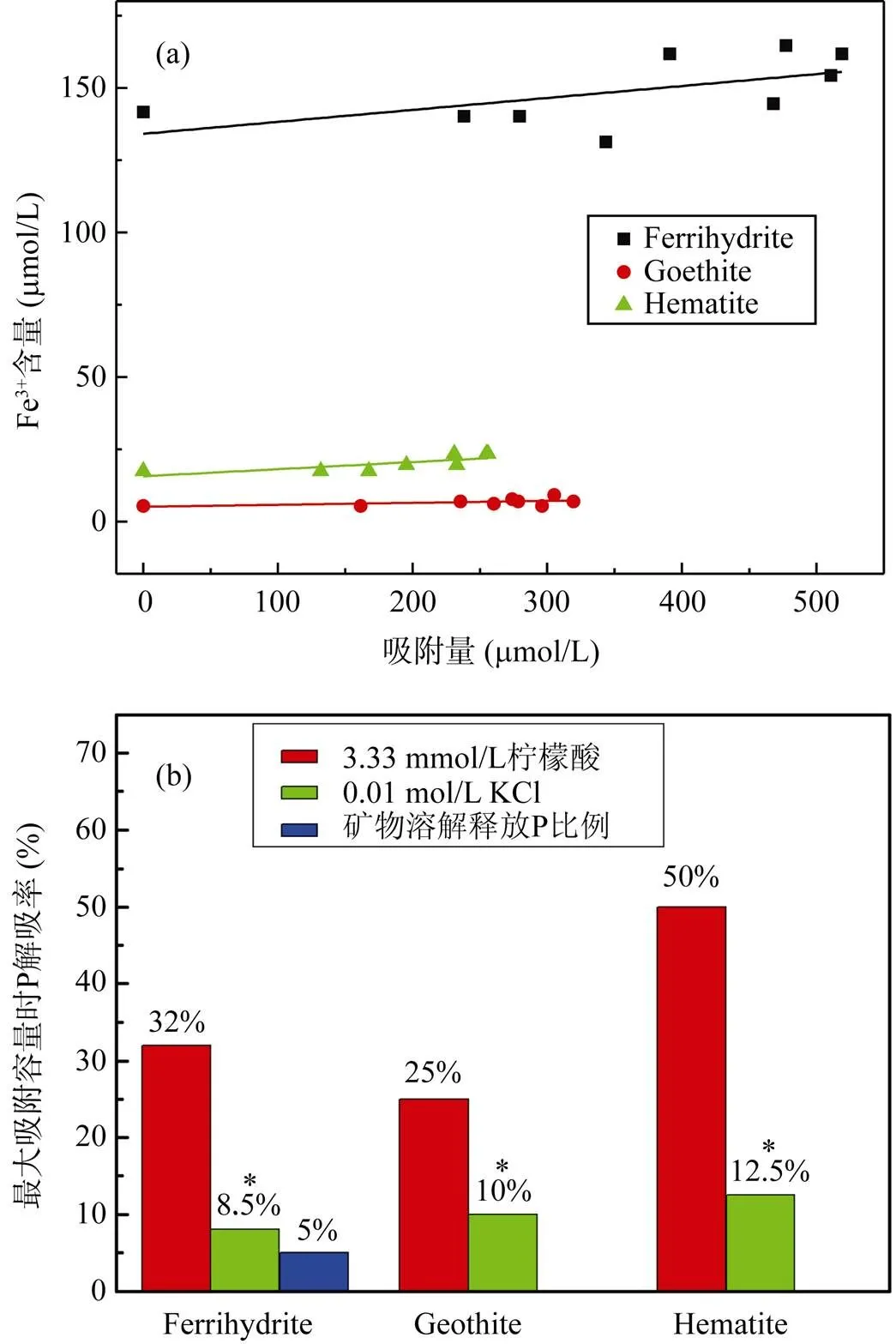
图10 柠檬酸解吸液中Fe3+含量随吸附量变化曲线(a)和最大吸附量时三种矿物在不同条件下的解吸率(b)
* 表示在0.01 < P < 0.05时, KCl解吸相对于柠檬酸解吸有显著性差异。
* means significant difference at 0.01 < P < 0.05 between KCl and citric acid desorption.
Fe oxide-P + L → Fe oxide-L + Paq(1)
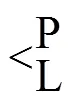
式中: L代表有机酸, Paq代表无机P。
3 结 论
(1)水铁矿是一种弱晶质的细小球形纳米颗粒, 结构中含有大量吸附水和一定量OH,且微孔发达; 针铁矿结构中含有大量OH; 赤铁矿结构中仅含少量H2O和OH。
(2)三种矿物都表现出初始快速反应和紧接的缓慢反应, 快速阶段三种矿物的速率常数相当, 慢速阶段速率常数水铁矿 > 针铁矿 > 赤铁矿, 且水铁矿存在持续的缓慢扩散反应过程。
(3)与针铁矿和赤铁矿相比, 水铁矿的吸附容量和OH–释放量更大且更符合F型等温线。水铁矿的特异性与它表面不饱和位点, 高表面位点密度和弱结晶特性密切相关。
(4)KCl介质中, 水铁矿是最难解吸的, 它的专性吸附比例最高。柠檬酸可以明显地促进铁(氢)氧化物释放P且诱导水铁矿溶解的效应更显著。
(5)与晶质氧化铁相比, 水铁矿结晶弱、微孔面积大, 表面位点密度更高, 不均一性大, 对P吸附量和专性吸附比例更高、吸附速率更快, 在一定的环境条件下对P的环境地球化学行为起着更重要和特殊的作用。
[1] 石元春. 20世纪中国学术大典: 农业科学[M]. 福州: 福建教育出版社, 2002: 228. Shi Yuan-chun. Chinese Academic Canon in the 20th Century: Agriculturals Sciences [M]. Fuzhou: Fujian Education Press, 2002: 228 (in Chinese).
[2] 苏德纯, 张福锁, 李国学. 磷-金属(Fe、Al)-有机酸三元复合体在植物磷营养中的作用[J]. 土壤通报, 2000, 31(4): 159-161.Su De-chun, Zhang Fu-suo, Li Guo-xue. Roles of phosphorus- metal(Fe or Al)-organic acid tri-complex on P nutrition of plants[J]. Chinese J Soil Sci, 2000,31(4): 159-161 (in Chinese with English abstract).
[3] McBride M B. Reactions controlling heavy metal solubility in soils[J]. Adv Soil Sci, 1989, 10: 1-56.
[4] Enwezor W O, Moore A W. Phosphorus status of some Nigerian soils[J]. Soil Sci, 1966, 102(5): 322-328.
[5] Shang C, Stewart J W B, Huang P M. pH effect on kinetics of adsorption of organic and inorganic phosphates by short-range ordered aluminum and iron precipitates[J]. Geoderma, 1992, 53(1/2): 1-14.
[6] Hayes K F, Papelis C, Leckie J O. Modeling ionic strength effects of anion adsorption at hydrous oxide/solution interface [J]. J Colloid Interface Sci, 1988, 125(2): 717-726.
[7] Arai Y, Sparks D L. ATR-FTIR spectroscopic investigation on phosphate adsorption mechanisms at the ferrihydrite-water interface [J]. J Colloid Interface Sci, 2001, 241(2): 317-326.
[8] Borggaard O K, Raben-Lange B, Gimsing A L, Strobel B W. Influence of humic substances on phosphate adsorption by aluminium and iron oxides [J]. Geoderma, 2005, 127(3/4): 270-279.
[9] Weng L P, van Riemsdijk W H, Hiemstra T. Humic nanoparticles at the oxide-water interface: Interactions with phosphate ion adsorption [J]. Environ Sci Technol, 2008, 42(23): 8747-8752.
[10] Nilsson N, Lövgren L, Sjöberg S. Phosphate complexation at the surface of goethite[J]. Chem Spec Bioavailab, 1992, 4: 121-130.
[11] Strauss R, Brümmer G W, Barrow N J. Effects of crystallinity of goethite: II. Rates of sorption and desorption of phosphate [J]. Eur J Soil Sci, 1997, 48(1): 101-114.
[12] Johnson S E, Loeppert R H. Role of organic acid in phosphate mobilization from iron oxide [J]. Soil Sci Soc Am J, 2006, 70(1): 222-234.
[13] Khare N, Martin J D, Hesterberg D. Phosphate bonding configuration on ferrihydrite based on molecular orbital calculations and XANES fingerprinting [J]. Geochim Cosmochim Acta, 2007, 71(18): 4405-4415.
[14] Antelo J, Fiol S, Pérez C, Mariño S, Arce F, Gondar D, López R. Analysis of phosphate adsorption onto ferrihydrite using the CD-MUSIC model [J]. J Colloid Interface Sci, 2010, 347(1): 112-119.
[15] 刘凡, 介晓磊, 贺纪正, 周代华, 徐风琳, 李学垣. 不同pH条件下针铁矿表面磷的配位形式及转化特点[J]. 土壤学报, 1997, 34(4): 368-374. Liu Fan, Jie Xiao-lei, He Ji-zheng, Zhou Dai-hua, Xu Feng-lin, Li Xue-yuan. Coordination forms and transformations of phosphate adsorbed by goethite surface on different pH [J]. Acta Pedol Sinica, 1997, 34(4): 368-374 (in Chinese with English abstract).
[16] Tejedor-Tejedor M I, Anderson M A. The protonation of phosphate on the surface of goethite as studied by CIR-FTIR and electrophoretic mobility [J]. Langmuir, 1990, 6(3): 602-611.
[17] Guzman G, Alcantara E, Barron V,Torrent J. Phytoavailability of phosphate adsorbed on ferrihydrite, hematite, and goethite [J]. Plant Soil, 1994, 159(2): 219-225.
[18] 刘凡, 李学垣, 徐风琳, 王代长, 荣艺. 鄂湘两省几种土壤中磷的有效性与氧化铁的类型[J]. 中国农业科学, 1995, 28(3): 49-57. Liu Fan, Li Xue-yuan, Xu Feng-lin, Wang Dai-zhang, Rong Yi. The effectiveness of phosphorus and types of iron oxides of several soils in Hubei and Hunan provinces [J]. Sci Agr Sinica, 1995, 28(3): 49-57 (in Chinese with English abstract).
[19] 王小明, 杨凯光, 孙世发, 徐剑, 李耀光, 刘凡, 冯雄汉. 水铁矿的结构、组成及环境地球化学行为[J]. 地学前缘, 2011, 18(2): 340-347.Wang Xiao-ming, Yang Kai-guang, Sun Shi-fa, Xu Jian, Li Yao-guang, Liu Fan, Feng Xiong-han. The structure and composition of ferrihydrite and its environmental geochemical behaviors[J]. Earth Sci Front, 2011, 18(2): 340-347 (in Chinese with English abstract).
[20] Wilson G V, Rhoton F E, Selim H M. Modeling the impact of ferrihydrite on adsorption-desorption of soil phosphorus [J]. Soil Sci, 2004, 169(4): 271-281.
[21] Rhoton F E, Bigham J M. Phosphate adsorption by ferrihydrite-amended soils [J]. J Environ Qual, 2005, 34(3): 890-896.
[22] Schwertmann U, Cornell R M. Iron Oxides in the Laboratory: Preparation and Characterization (2nd ed)[M]. Weinheim: Wiley-VCH, 1991: 1-137.
[23] Abou-Hassan A, Sandre O, Neveu S, Cabuil V. Synthesis of goethite by separation of the nucleation and growth processes of ferrihydrite nanoparticles using microfluidics [J]. Angew Chem Int Ed, 2009, 48(13): 2342-2345.
[24] Drits V A, Sakharov B A, Salyn A L, Manceau A. Structural model for ferrihydrite [J]. Clay Miner, 1993, 28(2): 185-207.
[25] Zhao J M, Huggins F E, Feng Z, Huffman G P. Ferrihydrite: Surface structure and its effects on phase transformation [J]. Clay Clay Miner, 1994, 42(6): 737-746.
[26] Jambor J L, Dutrizac J E. Occurrence and constitution of natural and synthetic ferrihydrite, a widespread iron oxyhydroxide [J]. Chem Rev, 1998, 98(7): 2549-2586.
[27] Arai Y, Sparks D L. Phosphate reaction dynamics in soils and soil components: A multiscale approach [J]. Adv Agron, 2007, 94: 135-178.
[28] Parfitt R L. Phosphate reactions with natural allophane, ferrihydrite and goethite [J]. J Soil Sci, 1989, 40(2): 359-369.
[29] Brantley S L, Kubicki J D, White A F. Kinetics of Water-Rock Interaction [M]. New York: Springer, 2007: 112.
[30] Willett I R, Chartres C J, Nguyen T T. Migration of phosphate into aggregated particles of ferrihydrite [J]. J Soil Sci, 1988, 39(2): 275-282.
[31] Celi L, de Luca G, Barberis E. Effects of interaction of organic and inorganic P with ferrihydrite and kaolinite-iron oxide systems on iron release [J]. Soil Sci, 2003, 168(7): 479-488.
[32] Koopal L K. Mineral hydroxides: From homogeneous to heterogeneous modeling [J]. Electrochim Acta, 1996, 41(14): 2293-2306.
[33] Hiemstra T, van Riemsdijk W H. A surface structural model for ferrihydrite I: Sites related to primary charge, molar mass, and mass density [J]. Geochim Cosmochim Acta, 2009, 73(15): 4423-4436.
[34] Salazar-Camacho C, Villalobos M. Goethite surface reactivity: III. Unifying arsenate adsorption behavior through a variable crystal face-Site density model [J]. Geochim Cosmochim Acta, 2010, 74(8): 2257-2280.
[35] Torrent J, Barron V, Schwertmann U. Phosphate adsorption and desorption by goethites differing in crystal morphology[J]. Soil Sci Soc Am J, 1990, 54(4): 1007-1012.
[36] Barron V, Herruzo M, Torrent J. Phosphate adsorption by aluminous hematite of different shapes [J]. Soil Sci Soc Am J, 1988, 52(3): 647-651.
[37] 绍兴华, 章永松, 林咸勇, 都韶婷, 于承艳. 三种铁氧化物的磷吸附解吸特性以及与磷吸附饱和度的关系[J]. 植物营养与肥料学报, 2006, 12(2): 208-212. Shao Xing-hua, Zhang Yong-song, Lin Xian-yong, Du Shao-ting, Yu Cheng-yan. Phosphorus adsorption and desorption properties of three synthetic iron oxides and their relation to phosphorus adsorption saturation [J]. Plant Nutr Fert Sci, 2006, 12(2): 208-212 (in Chinese with English abstract).
[38] Stumm W, Fürrer G. The dissolution of oxides and aluminum silicates: Examples of surface-coordination-controlled kinetics[M]//Stumm W. Aquatic Surface Chemistry: Chemical Processes at the Particle-Water Interface. New York: John Wiley & Sons, 1987: 197-219.
The P adsorption-desorption characteristics on ferrihydrite and crystalline Fe oxides suspension
WANG Xiao-ming, SUN Shi-fa, LIU Fan, TAN Wen-feng, HU Hong-qing and FENG Xiong-han*
(Key Laboratory of Subtropical Agriculture Resource and Environment, Ministry of Agriculture, College of Resources and Environment, Huazhong Agricultural University, Wuhan 430070, China)
The retention and release of P onto iron oxides influence and determine the bioavailability and eutrophication of P. We study the adsorption-desorption characteristics of P onto ferrihydrite and explore the relevant mechanisms compared with two common crystalline Fe oxides (goethite and hematite), using the techniques of X-ray diffraction (XRD), transmission electron microscopy (TEM), thermal gravimetric analysis (TGA), porosity structure analysis and the methods of kinetics and adsorption-desorption equilibrium. Results show a fast initial adsorption took place in a few minutes followed by a slow reaction process, both of which can be well fitted by first order kinetics. During the reaction, the release of OH–lags behind P adsorption and increases step by step, indicating that P adsorption may go through complexation from outer-sphere to inner-sphere, from monodentate to bidentate coordination. Compared with goethite and hematite, ferrihydrite has higher amount of P adsorption and OH–release, faster slow reaction process, and exhibits slow diffusion reaction stage. The maximum adsorption capacities are in order of: ferrihydrite (4.36 μmol/m2) >> goethite (2.62 μmol/m2) > hematite (2.28 μmol/m2); adsorption isotherms of goethite and hematite agree well with Langmuir model while ferrihydrite exhibits an adsorption isotherm of Freundlich type. Desorption percentage of adsorbed P by neutral electrolyte at maximum adsorption capacities is in order of: ferrihydrite (8.5%) < goethite (10%) < hematite (12.5%). Relatively, citric acid significantly promotes the desorption reaction. Desorption percentages at the maximum adsorption capacity are in order of: goethite (25%) < ferrihydrite (32%) < hematite (50%).
iron (hydr)oxides; ferrihydrite; goethite; hematite; P; adsorption-desorption
P595
A
0379-1726(2012)01-0089-10
2011-02-25;
2011-06-01;
2011-06-03
国家自然科学基金重大项目(30890132, 30890133); 国家自然科学基金(41171197); 国家大学生创新性试验计划项目(091050404)
王小明(1985–), 男, 博士研究生, 环境土壤化学研究方向。E-mail: wangxm338@163.com
FENG Xiong-han, E-mail: fxh73@mail.hzau.edu.cn; Tel: +86-27-87280271
