HPLC测定蒙成药塔日努苏木朱尔散剂中大黄酚的含量
2012-06-06周晓明王晓琴
周晓明 王晓琴 李 波
(1.内蒙古呼伦贝尔市食品药品检验所,内蒙古 呼伦贝尔 021008;2.内蒙古医学院药学院,内蒙古 呼和浩特 010110)
塔日努苏木朱尔散剂是由波叶大黄、狼毒、白矾3味药组成的蒙药复方制剂,具有清热燥湿、杀虫止痒的功效,用于治疗皮炎、牛皮癣、湿疹、皮肤瘙痒。其中波叶大黄为方中主药。波叶大黄(Rheum undulatum),又名山大黄,为蓼科大黄属药用植物,属呼伦贝尔市地产蒙药材,有破积滞,行瘀血的作用,可用于痈肿疔毒,跌打瘀痛,口疮糜烂等症[1]。其根及根茎主要含有以大黄酚和大黄素为苷元的结合型蒽醌和游离型蒽醌;其中,游离型蒽醌中主要为大黄酚和大黄素甲醚[2]。为了保证产品内在质量,本文选择大黄酚作为质量控制指标,参照“中国药典”2010年版一部“大黄”项下的含量测定方法[3],采用高效液相色谱法建立了大黄酚的含量测定方法。并经过方法学考察及阴性对照实验,表明该方法重现性好,专属性强,方中其它组分对大黄酚的测定无干扰,可作为该复方制剂的质控方法。
1 仪器与试药
1.1 仪器:岛津LC-2010A HT高效液相色谱仪;CBM-20A型控制器;SPD-10Avp型检测器,LC solution色谱工作站。Sartorius Bp121S(0.1mg)、Bp211D(0.01mg)电子天平。
1.2 试剂与试药:大黄酚对照品由中国药品生物制品检定所提供(批号110796-2003120);甲醇为色谱纯;水为高纯水,其它试剂均为分析纯。蒙成药塔日努苏木朱尔散剂由呼伦贝尔市新巴尔虎右旗蒙医医院提供。
2 方法与结果
2.1 色谱条件:Diamonsil RP-C18色谱柱(250× 4.6mm,5μm);流动相为甲醇 -0.1%磷酸(85﹕ 15);柱温30℃;检测波长254nm;流速1mL·min-1;进样量10μL。
2.2 溶液的制备
2.2.1 对照品溶液的制备:精密称取大黄酚对照品适量,加甲醇制成每1mL含32.16μg的溶液,即得。
2.2.2 供试品溶液的制备:取本品粉末约1g,精密称定,置具塞锥形瓶中,精密加入甲醇25mL,称定重量,加热回流1h,放冷,再称定重量,用甲醇补足减失的重量,摇匀,滤过。精密量取续滤液5mL,置烧瓶中,挥去溶剂,加8%盐酸溶液10mL,超声处理2min,再加三氯甲烷10mL,加热回流1h,放冷,置分液漏斗中,用少量三氯甲烷洗涤容器,并入分液漏斗中,分取三氯甲烷层,酸液再用三氯甲烷提取3次,每次10mL,合并三氯甲烷液,减压回收溶剂至干,残渣加甲醇使溶解,转移至10mL量瓶中,加甲醇至刻度,摇匀,用0.45μm滤膜滤过,取续滤液,即得。
2.2.3 阴性空白对照溶液的制备:按处方比例并以相同工艺制备不含波叶大黄的空白样品,按供试品溶液制备法制得阴性对照溶液。
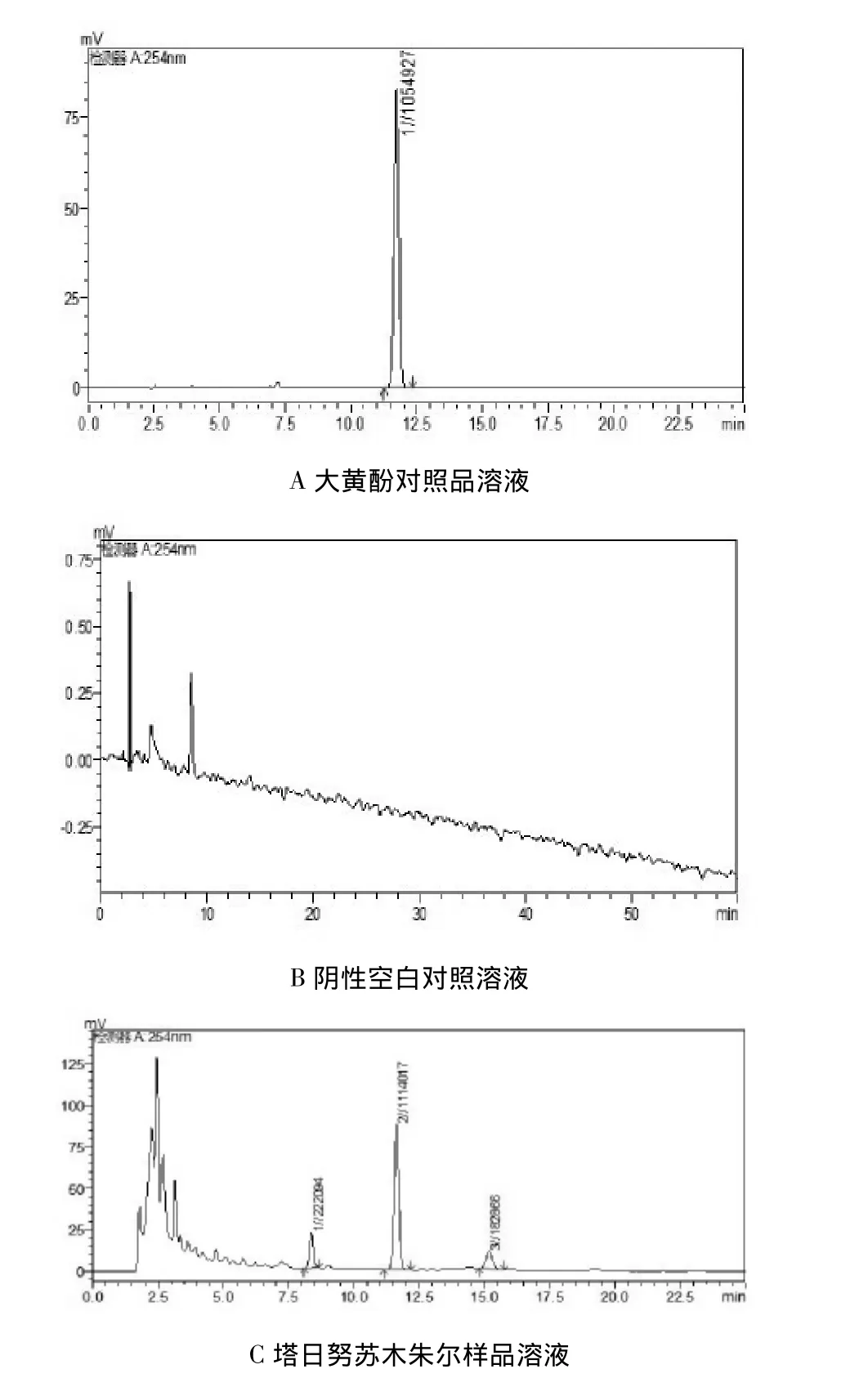
图1 HPLC色谱图
2.3 空白阴性实验:分别精密吸取对照品溶液、阴性空白对照溶液、供试品溶液各10μL,注入液相色谱仪,记录色谱图,见图1。结果阴性空白对照溶液色谱图中在与大黄酚对照品及供试品色谱图相对应的保留时间处无色谱峰出现,表明处方中其它组分对大黄酚的测定无干扰,在此条件下,大黄酚和相邻成分完全分离。
2.4 线性关系考察:精密称取大黄酚对照品约10mg,置25mL量瓶中,加甲醇溶解并稀释至刻度;精密量取0.5、2、3、4、5mL置25mL量瓶中,加甲醇稀释到刻度,摇匀,各取10μL进样,按上述色谱条件测定,以峰面积对进样量进行回归分析,结果大黄酚在 0.00804~0.0804μg·mL-1浓度范围内与封面积呈良好的线性关系。回归方程为Y=518724X -1711.3,r=0.9995。
2.5 稳定性试验:取样品含量测定项下制备的供试品溶液1份,分别于0、4、8、12、16、24h 进样测定,结果大黄酚峰面积基本不变,RSD为0.27%(n=6)。试验结果表明,大黄酚在24h内稳定性良好。
2.6 精密度试验:精密吸取对照品溶液10μL,重复进样6次,测定峰面积积分值,RSD为0.4%。试验结果表明仪器精密度良好。
2.7 重现性试验:取同一批号样品6份,约1.0g,精密称定,按供试品溶液的制备方法制得样品溶液,按“2.1”项下的色谱条件分析,测得平均含量为1.457mg·g-1,RSD为1.8%,实验结果表明该方法重现性良好。
2.8 加样回收率试验:精密称取已知大黄酚含量(含量为1.457mg·g-1)的样品 9份,每份约 0.6g,分别精密加入浓度为0.143m·mL-1的大黄酚对照品甲醇溶液5mL、10mL、15mL,制成低、中、高3种浓度的溶液各3份,按供试品溶液的制备方法制得样品溶液,按“2.1”项下的色谱条件分析,计算回收率。结果平均回收率为100.4%,RSD%为1.96%。结果见表1。
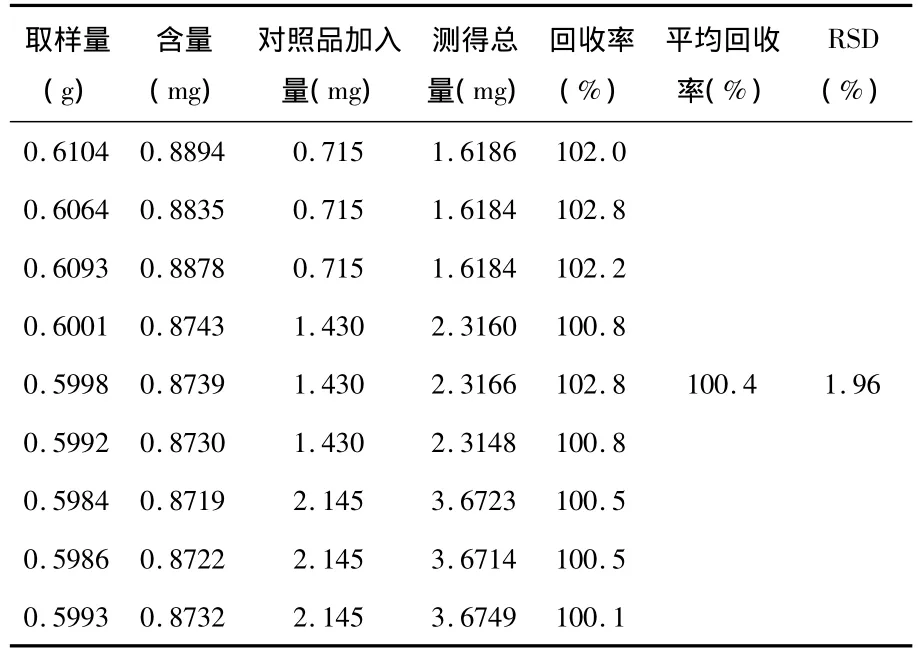
表1 加样回收率试验结果
2.9 样品的含量测定:取本品3个批号共6份,各约1.2g,按供试品溶液的制备方法处理,分别精密吸取供试品溶液与对照溶液各10μL注入液相色谱仪,按外标法计算含量。三批样品的测定结果见表2。
3 讨论
3.1 检测波长的选择:取大黄酚对照品溶液,于紫外-可见分光光度计上,自190nm-600nm做光谱扫描,结果大黄酚在255.5nm和428.5nm处有最大吸收,在428.5nm波长下,基线不稳,结合“中国药典”一部大黄酚含量测定项下的测定方法,选择254nm作为检测波长。
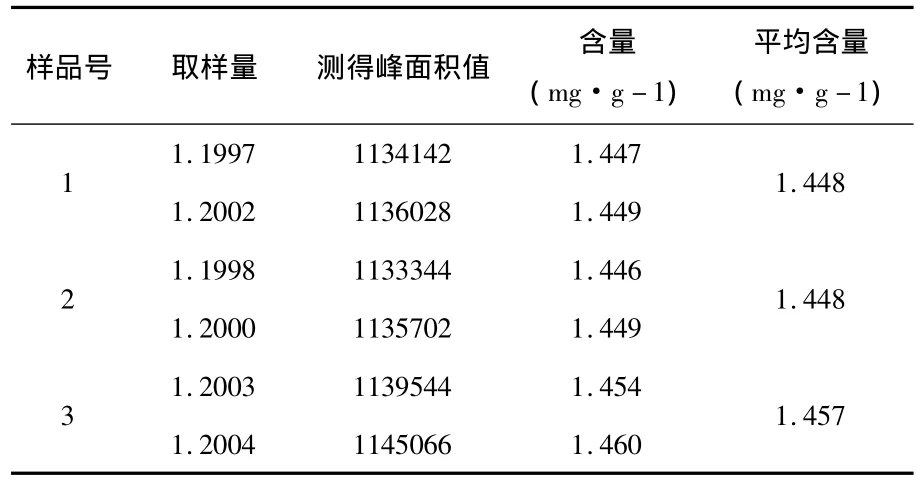
表2 样品中大黄酚的含量测定结果
3.2 提取条件的选择:参照《“中国药典”2010年版一部“大黄”项下的含量测定方法,考虑到大黄酚易溶于三氯甲烷等低极性有机溶剂,故选择用甲醇加热回流提取,然后用三氯甲烷萃取。实验中考察了提取3次、4次、5次、6次等不同提取次数对提取效率的影响,结果表明提取4次后供试品中大黄酚的含量最高,故将提取次数定为4次。
本实验精密度及回收率试验结果表明,该方法具有较好的准确度和重现性,可作为该制剂的含量测定方法。
[1]国家中医药管理局中华本草编委会.中华本草[M],第六卷.上海:上海科学技术出版社,1999:2705.
[2]肖培根.大黄属的植物亲缘关系、化学成分与疗效间联系性的初步研究[J].药学学报,1980,(1):33-39.
[3]国家药典委员会.中华人民共和国药典2010版(一部)[S].北京:中国医药科技出版社,2010:22.
