Leber's遗传性视神经病叠加综合征一家系
2012-05-30庄淑流刘利娟
童 绎 庄淑流 刘利娟
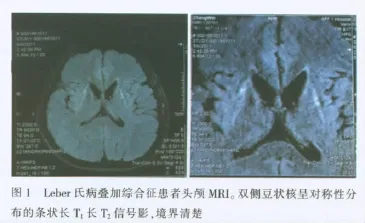
笔者早年对Leber氏病(Leber hereditary optic neuropathy,LHON)的mtDNA 11778位点突变家系进行研究时,发现了3个家系,这3个家系除有典型女性垂直传递及家族中有2人以上患视神经萎缩外,还有不同程度的神经系统症状,各家系患者及其母系中均未发现血mtDNA 11778位点突变,其他位点未进行检查〔1〕。复习文献后认为,临床表现符合LHON+,即叠加综合征。近年来对其中一家系,除先证者外的其他成员和135名正常对照者进行mtDNA T3866C突变筛查,结果这家系所有母系成员均携带该突变,非母系成员和135名正常对照均不携带此突变。以上结果提示,T3866C突变可能是与LHON相关的mtDNA突变〔2〕。因为该家系有明显的神经系统并发症,临床诊断为LHON+,即Leber氏病叠加综合征,考虑其发病可能与ND1基因T3866C位点突变有关。此外,经神经内科会诊发现Ⅳ13有典型的痉挛性肌张力障碍,纠正了以前四肢畸形跛行的诊断。Ⅳ13头颅MRI显示:双侧豆状核呈对称性分布的条状长T1长T2信号影,境界清楚(图1)。
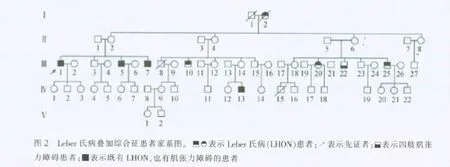
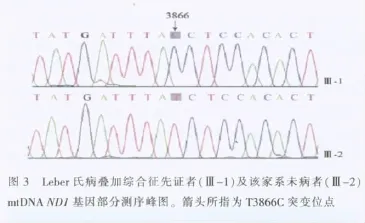
今特报告如下。先证者Ⅲ-1,男,58岁,福建连江县人,30岁时双眼先后视力逐渐减退,曾至医院诊断为视神经炎,其后发展为视神经萎缩,经治疗无好转。眼部检查:双眼视力数指/眼前,双瞳孔大小及对光反应尚正常,双眼底见视盘色苍白,双VEP-P100潜时延长,振幅降低。有智力迟钝,言语明显含糊不清,四肢肌张力障碍。对家系中5代61人(图2)有些成员进行系统的临床检查。从图中可见,LHON同时有肌张力障碍4例,仅有视神经萎缩者4例,1例有肌张力障碍。言语含糊不清者3例,智力不同程度减退4例;为一典型母系遗传方式。发病年龄10岁至44岁。男性发病6人,女性2人,外显率为29.6%。该家系成员无其他临床症状。先证者(Ⅲ-1)及该家系未病者(Ⅲ-2)mtDNA基因部分测序峰图见图3。
讨论
Leber氏病(Leber hereditary optic neuropathy,LHON)家系中有多个成员合并严重神经系统异常,则称为LHON+,即Leber氏病叠加综合征。亦有认为除视神经病变外,尚可有震颤、肌张力障碍、运动失调、小脑性共济失调、精神异常、骨骼异常、脊髓病变、急性婴儿期脑性癫痫、儿童期致死性脑病等,影像学示基底核病变等〔3-4〕。
Newman 首先用 LHON+表示 Leber氏病叠加综合征〔5〕。Nikoskelainen等〔5〕对一组有神经系统受累的LHON 46例重新复查,计男性38例,女性8例,根据mtDNA分析将其分为3组:一组27例为11778位点突变,一组9例为3460位点突变,第3组10例无原发性位点突变,但发现在ND5的12811、13967,ND2的4732和ND5的13637位点存在突变。Johns等报告15257位点突变见于并发有脊髓和周围神经病变者〔5〕。本文所报告家系可能为ND1的T3866C位点突变,尚未见文献报告。唐氏等〔6〕认为LHON+除以视力减退,色觉障碍起病的视神经病变外,还可发现痉挛性肌张力障碍,共济失调、周围神经病、脊髓病等其他神经系统损害,部分患者甚至可伴发“自身免疫病”样,或多发性硬化样临床症状,均称为LHON+。
有文献提到LHON+这些家系基因突变形式通常与典型的LHON基因突变有不同之处〔3〕,这与前述3个家系11778突变位点均为阴性的结果相符合〔1〕。本家系具有LHON表现和(或)四肢肌张力障碍、言语不清、智力减退的患者只存在于母系成员中,提示mtDNA突变可能为其发病的分子基础。通过对先证者的线粒体全基因组分析,未发现LHON常见三个原发位点突变,但发现有ND1基因T3866C位点的突变,我们认为该突变位点可能即为该病的相关线粒体基因突变。然而该系家庭成员还具有不完全外显、发病年龄不同、视力损害程度不等的特点,说明T3866C突变本身可能不足以导致此病,其他如核修饰基因、环境因素等在发病中也起到一定的促进作用〔2〕。
Larssor等〔5〕报告1例有单侧肢体震颤和肌强直者伴有11778位点突变,1年后在36岁时发生视神经受累,头颅MRI显示明显的双侧豆状核病变,类似文献亦有报告,与本家系所见1例相同。LHON叠加综合征行头颅MRI检查有一定临床诊断价值,有助于与其他颅内疾病进行鉴别或排除。
上海华山医院唐氏等〔6〕报告1例男性37岁患者,主诉视力进行性下降4年,行走困难1年半。首先发生辨色困难,后出现视力下降,双侧视神经萎缩,双VEP-P100潜伏期明显延长,伴波幅明显降低,其后尚有脊髓病样表现,脑脊液蛋白增高,寡克隆区带(oligoclonal band,OB)阳性,IgG 指数10.92,血清视神经脊髓炎(NMO)抗体阴性。经激素治疗后症状能部分缓解,临床上一度首先考虑视神经脊髓炎(Neuromyelitis Optica,NMO)或进展性多发性硬化(multiple sclerosis,MS)的诊断,但查NMO抗体阴性,且患者头颅、颈髓、胸髓MRI示颈髓、胸髓上段及中段萎缩,缺乏NMO与MS典型影像学的表现,患者血液线粒体DNA检测发现有11778位点突变,最终诊断为伴多发性硬化样表现的Leber病叠加综合征。芬兰Nikoskelainen等对46例LHON患者进行再检查发现,仅有2例表现为多发性硬化样综合征,该2例患者头颅MRI均显示有脑室周围白质病变〔5〕。唐氏等〔6〕认为双视力下降,伴有视神经病变,其后有其他全身部位受累或出现自身免疫病变样表现,且病程进展缓慢的患者,应考虑行外周血线粒体mtDNA原发性位点等检测。
[1] 童 绎,高静娟.Leber病伴神经系统并发症及视力恢复与线粒体DNA11778 位点突变相关研究[J].中华眼底病杂志,1997,13(3):183-184.
[2] 刘 燕,庄淑流,童 绎,等.线粒体ND1基因T3866C突变可能是Leber’s遗传性视神经病和四肢畸形跛行相关的突变[J].遗传,2010,32(2):141-147.
[3] Neil R Miller, Nancy J Newman, Valérie Biousse, 等.Walsh and Hoyt精编临床神经眼科学[M].张晓君,魏文斌,译.北京:科学出版社,2009:234.
[4] 童 绎,魏世辉,游思维.视路疾病基础与临床进展[M].北京:人民卫生出版社,2011:342.
[5] Nikoskelainen EK, Marttila RJ, Huoponen K, et al.Leber’s“plus”neurological abnormalities in patients with Leber’s hereditary optic neuropathy[J].J Neurol Neurosurg Psychiatry, 1995,59(2): 160-164.
[6] 唐兴华,吴洵昳,章 悦,等.伴有自身免疫病样表现的Leber遗传性视神经病叠加综合征一例[J].中华神经科杂志,2010,43(1):74-76.
