N-取代苯基-2-(4-取代苯基)环丙烷-1-甲酸乙酯-1-酰胺的设计、合成及抗肿瘤活性
2012-01-30耿冬平第二军医大学药学院药物化学教研室上海200433
陈 焕,耿冬平,李 科(第二军医大学药学院药物化学教研室,上海200433)
肿瘤特别是恶性肿瘤严重威胁人类健康。据世界卫生组织调查,由恶性肿瘤引起的死亡每年约占所有疾病死亡人数的13%,并且呈逐年增长趋势[1]。恶性肿瘤预后差、造成患者痛苦、治疗费用高,不仅严重影响人们身心健康,而且给家庭、社会造成沉重经济负担,成为影响社会和谐发展的不容忽视的因素。临床化学药物治疗是抗肿瘤的主要手段之一,因此,研发抗肿瘤药物是国内外药物研究的重要方向,也是我国“十一五”、“十二五”连续规划的重点药物研发领域。
从中药中获取有效成分,进而对其进行化学、药理和临床研究,是现代科技对中药的巨大发展之一[2]。周大铮等从香榧假皮中提取分离得到了具有一定抗肿瘤活性的单体2-芳基-4-苄基四氢呋喃木脂素TG-10[3,4](图1)。为了获取更高抗肿瘤活性的TG-10类衍生物,笔者对TG-10进行了结构改造和结构修饰,获得了比较满意的结果[5~7]。构效关系研究发现,TG-10的呋喃环非药效必需基团且该环合成复杂。因此,笔者采用微波反应,经缩合、环化、水解及酰胺化等反应设计、合成了20个未见文献报道的反式构型的N-取代苯基-2-(4-取代苯基)环丙烷-1-甲酸乙酯-1-酰胺系列化合物,目标化合物的结构通过1H NMR和MS确定。采用MTT法筛选体外抗肿瘤活性。实验结果显示,所合成的目标化合物具有较好的抗肿瘤活性。其中5b对A549细胞的IC50值达到6.8 μM,具有进一步研究的价值。微波反应实验结果与文献报道[8]相对比,在保证收率的同时,节约了原材料和防护了环境污染。合成路线见图2。

图1 TG-10的结构

图2 目标化合物的合成路线
1 实验部分
1.1 仪器和材料 熔点由RY-2型熔点仪进行测定,温度未经校正;核磁共振谱由Bruker AC-300P型仪器测定,TMS为内标,溶剂为DMSO;质谱由Finigan LCQ Deca XP MAX质谱仪测定;层析柱采用青岛海洋化工厂生产的HSGF254型硅胶H(10~40 μM);所用试剂均为分析纯试剂。
1.2 试剂干燥 无水CH2Cl2:CH2Cl2中加入CaH2(5 g/L),回流5 h,蒸馏后分子筛(4Å)保存;无水三乙胺:三乙胺中加入CaH2(5 g/L),回流16 h,蒸馏后分子筛(4Å)保存;无水 DMSO:DMSO中加入CaH2(5 g/L),回流5 h,蒸馏后分子筛(4Å)保存。
1.3 目标化合物的合成
1.3.1 4-氯苯基次甲基丙二酸二乙酯的制备 4-氯苯甲醛(14.57 g,0.1 mol)、丙二酸二乙酯(16.81 g,0.105 mol)、冰乙酸(3 ml,5.0 mmol)、哌啶(1.8 ml,3 mmol)、硅胶(1 g)依次加入干燥的甲苯(200 ml)中,搅拌回流反应12 h,分馏生成的水。减压蒸去甲苯,残留物用二氯甲烷(200 ml)和水(200 ml)分别萃取3次,合并有机相,减压蒸去二氯甲烷,残留固体用乙酸乙酯∶石油醚=2∶1(v/v)结晶。滤取结晶,真空干燥,得4-氯苯基次甲基丙二酸二乙酯。称重得27.05 g(95.0 mmol),收率95%。MP: 33.2~34.5℃。1H NMR(DMSO-d6,300 MHz):δ 8.15(s,1H),7.34(s,2H),6.56(s,2H),4.32~4.26(m,4H),1.21~1.16(m,6H)。
1.3.2 4-三氟甲基苯基次甲基丙二酸二乙酯的制备 4-三氟甲基苯甲醛(18.81 g,0.1 mol)、丙二酸二乙酯(16.81 g,0.105 mol)、冰乙酸(3 ml,5.0 mmol)、哌啶(1.8 ml,3 mmol)、硅胶(1 g))依次加入干燥的甲苯(200 ml)中,搅拌回流反应12 h,分馏生成的水。减压蒸去甲苯,残留物用二氯甲烷(200 ml)和水(200 ml)分别萃取3次,合并有机相,减压蒸去二氯甲烷,残留固体用乙酸乙酯∶石油醚=2∶1(v/v)溶解,置冰箱中结晶。滤取结晶,真空干燥,得4-三氟甲基苯基次甲基丙二酸二乙酯。称重得29.6 g(93.0 mmol),收率93%。MP:51.3~52.5℃。1H NMR(DMSO-d6,300 MHz):δ 8.35(s,1H),7.65(s,2H),7.43(s,2H),4.39~4.34 (m,4H),0.67~0.58(m,6H)。
1.3.3 2-(4-氯苯基)环丙烷-1,1-二甲酸二乙酯的制备 NaH(2.38 g,99 mmol)加入到无水DMSO(200 ml),搅拌溶解,分批加入三甲基碘化亚砜(21.78 g,99 mmol),室温搅拌至无气体放出。加入4-氯苯基次甲基丙二酸二乙酯(25.6 g,90 mmol);于60℃、400 W微波反应器中反应10 min。反应结束后反应液用二氯甲烷(200 ml)和水(300 ml)各萃取3次,合并有机相,减压蒸去二氯甲烷,残留固体用乙酸乙酯∶石油醚=2∶1(v/v)重结晶。过滤晶体后干燥,得4-氯苯基环丙烷丙二酸二乙酯,称重得25.37 g,收率95%。MP:77.7~78.5℃。1H NMR(DMSO-d6,300 MHz):δ 7.37(s,2H),6.54(s,2H),4.34~4.30 (m,4H),3.10~3.04(t,J=8.7 Hz,1H),2.03~2.01(m,1H),1.64~1.59(m,1H),0.77~0.72 (m,6H)。
1.3.4 2-(4-三氟甲基苯基)环丙烷二甲酸二乙酯的制备 NaH(2.38 g,99 mmol)加入到无水DMSO(200 ml),搅拌溶解,分批加入三甲基碘化亚砜(21.78 g,99 mmol),室温搅拌至无气体放出。加入4-三氟甲基苯基次甲基丙二酸二乙酯(25.6 g,90 mmol);60℃、400 W微波反应器中反应10 min。反应结束后反应液用二氯甲烷(200 ml)和水(300 ml)各萃取3次,合并有机相,减压蒸除溶剂。残留物用乙酸乙酯∶石油醚=2∶1(v/v)重结晶。过滤晶体后干燥,得4-三氟甲基苯基环丙烷丙二酸二乙酯,称重到27.65 g,收率93%。MP:125.5~126.1℃,1H NMR(DMSO-d6,300 MHz):δ 7.68(s,2H),7.47(s,2H),4.36~4.30 (m,4H),3.23~3.18(t,J=8.4 Hz,1H),2.16~2.12(m,1H),1.71~1.66(m,1H),0.76~0.71 (m,6H)。
1.3.5 2-(4-氯苯基)环丙烷-1,1-二甲酸单乙酯的制备 2-(4-氯苯基)环丙烷-1,1-二甲酸二乙酯(23.74 g,80 mmol)、乙醇∶水=10∶1(v/v)混合液(200 ml)和KOH(4.71 g,84 mmol),于30℃搅拌反应3~4 h,然后先用3%HCl调pH至7,再加水至总溶液的2倍,静置过夜,有大量白色固体析出,滤出固体,真空烘箱干燥,得2-(4-氯苯基)环丙烷-1,1-二甲酸单乙酯,19.4 g,收率90%。MP:98.6~99.4℃。1H NMR (DMSO-d6,300 MHz):δ 12.02(s,1H),7.39(s,2H),6.57(s,2H),4.32~4.28(m,2H),3.11~3.07(t,J=8.7 Hz,1H),2.23~2.19(m,1H),1.60~1.56(m,1H),0.76~0.73(m,3H)。
1.3.6 2-(4-三氟甲基苯基)环丙烷二甲酸单乙酯的制备 2-(4-氯苯基)环丙烷-1,1-二甲酸二乙酯(26.40 g,80 mmol)、乙醇∶水=10∶1(v/v)混合液(200 ml)和KOH(4.71 g,84 mmol),于30℃搅拌反应3~4 h,然后先用3%HCl调pH至7,再加水至总溶液的2倍,静置过夜,有大量白色固体析出,滤出固体,真空烘箱干燥,得2-(4-三氟甲基苯基)环丙烷二甲酸单乙酯,21.5 g,收率87%。MP:134.8~136.1℃。1H NMR(DMSO-d6,300 MHz):δ 11.63(s,1H),7.71(s,2H),7.45(s,2H),4.33~4.29(m,2H),3.21~3.17(t,J=8.4 Hz,1H),2.17~2.11(m,1H),1.70~1.66(m,1H),0.75~0.70(m,3H)。
1.3.7 (反式)N-苯基-2-(4-氯苯基)环丙烷-1-甲酸乙酯-1-甲酰胺(5a)的制备 2-(4-氯苯基)环丙烷-1,1-二甲酸单乙酯(1.34 g,5 mmol)、无水二氯甲烷(20 ml)和草酰氯(0.48 ml,5.05 mmol),室温搅拌反应6 h,反应结束后减压蒸去反应液,得酰氯;苯胺(0.54 ml,6 mmol)溶于无水二氯甲烷(10 ml)中,再加入无水吡啶(1.05 ml,15 mmol),冰浴冷至0℃,搅拌下滴入酰氯,滴毕保温30 min后常温反应过夜。先用3% HCl调pH至7.0,再用二氯甲烷和水萃取3次,合并有机相,用2%碱水洗涤3次,干燥后减压蒸馏有机相,残留物以乙酸乙酯:石油醚(v/v)为展开剂,经柱层析梯度洗脱(0%~3%;3%~6%;6%~10%)得到(反式)N-苯基-2-(4-氯苯基)环丙烷-1-甲酸乙酯-1-甲酰胺。0.56 g,收率65%。
同法制得5b~5j,化合物的理化性质及谱图数据见表1。
1.3.8 (反式)N-苯基-2-(4-三氟甲基苯基)环丙烷-1-甲酸乙酯-1-甲酰胺(6a)的制备 2-(4-三氟甲基苯基)环丙烷二甲酸单乙酯(1.51 g,5 mmol)、无水二氯甲烷(20 ml)和草酰氯(0.48 ml,5.05 mmol),室温搅拌反应6 h,反应结束后减压蒸去反应液,得酰氯;苯胺(0.54 ml,6 mmol)、无水二氯甲烷(10 ml)和无水吡啶(1.05 ml,15 mmol),冰浴冷至0℃,搅拌下滴入酰氯,滴毕保温30 min后常温反应过夜。先用3%HCl调pH至7.0,合并有机相,用2%,干燥后减压蒸馏有机相,残留物以乙酸乙酯∶石油醚(v/v)为展开剂,经柱层析梯度洗脱(0%~3%;3%~6%;6%~10%)得到反式结构N-苯基-2-(4-三氟甲基苯基)环丙烷-1-甲酸乙酯-1-甲酰胺。0.70 g,收率74.1%。
所得目标化合物的纯度经高效液相检测,使用Agilent 1100 series LC system,柱子规格为4.6 mm ×150 mm,填充物为C8;流动相为甲醇∶水=7∶3 (v/v);流速为1.0 ml/min;每次进样体20 μl;在254 nm紫外监控。所有化合物的纯度>95%。
同法制得6b~6j,化合物的理化性质及谱图数据见表1。
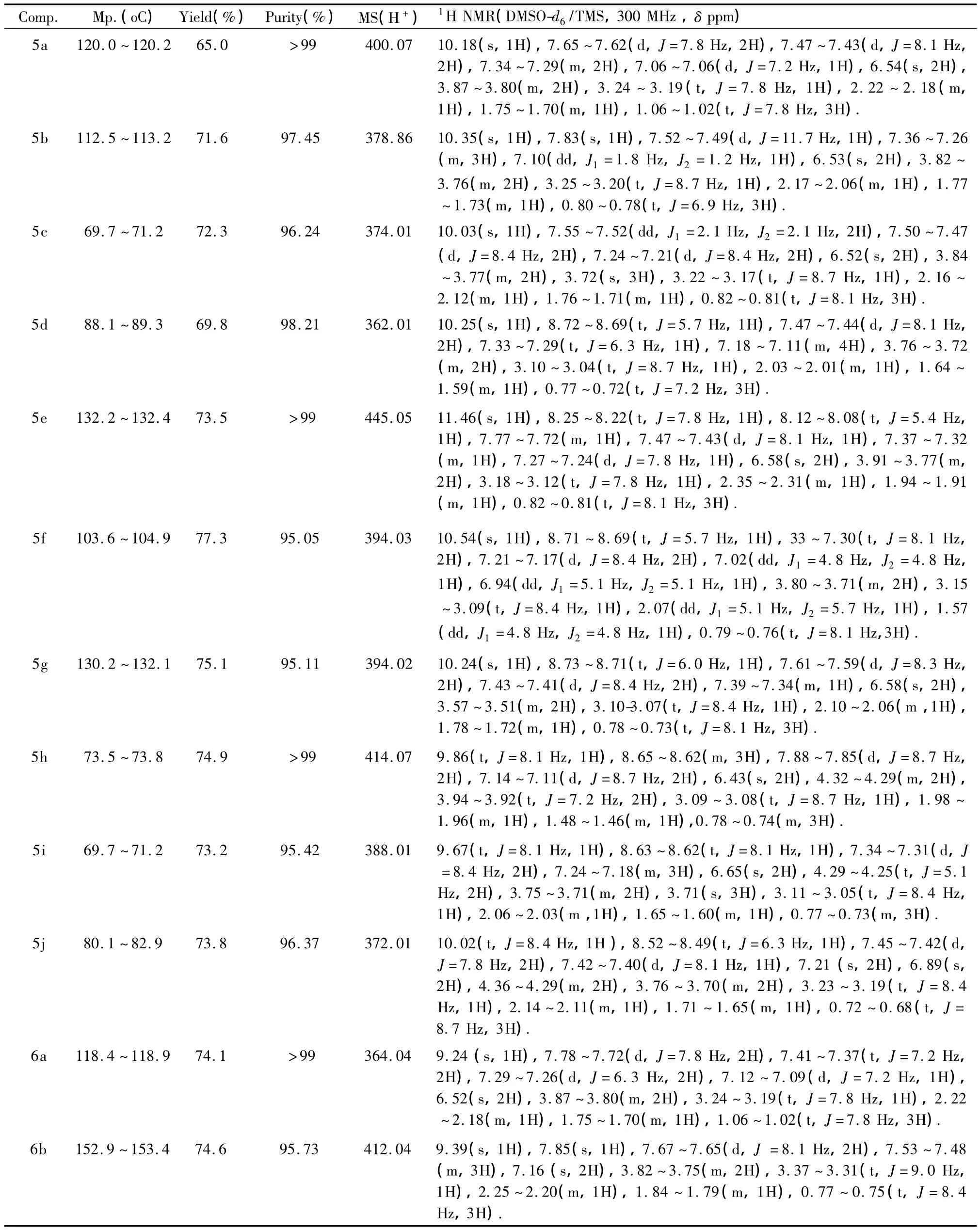
表1 目标化合物的理化性质及谱图数据
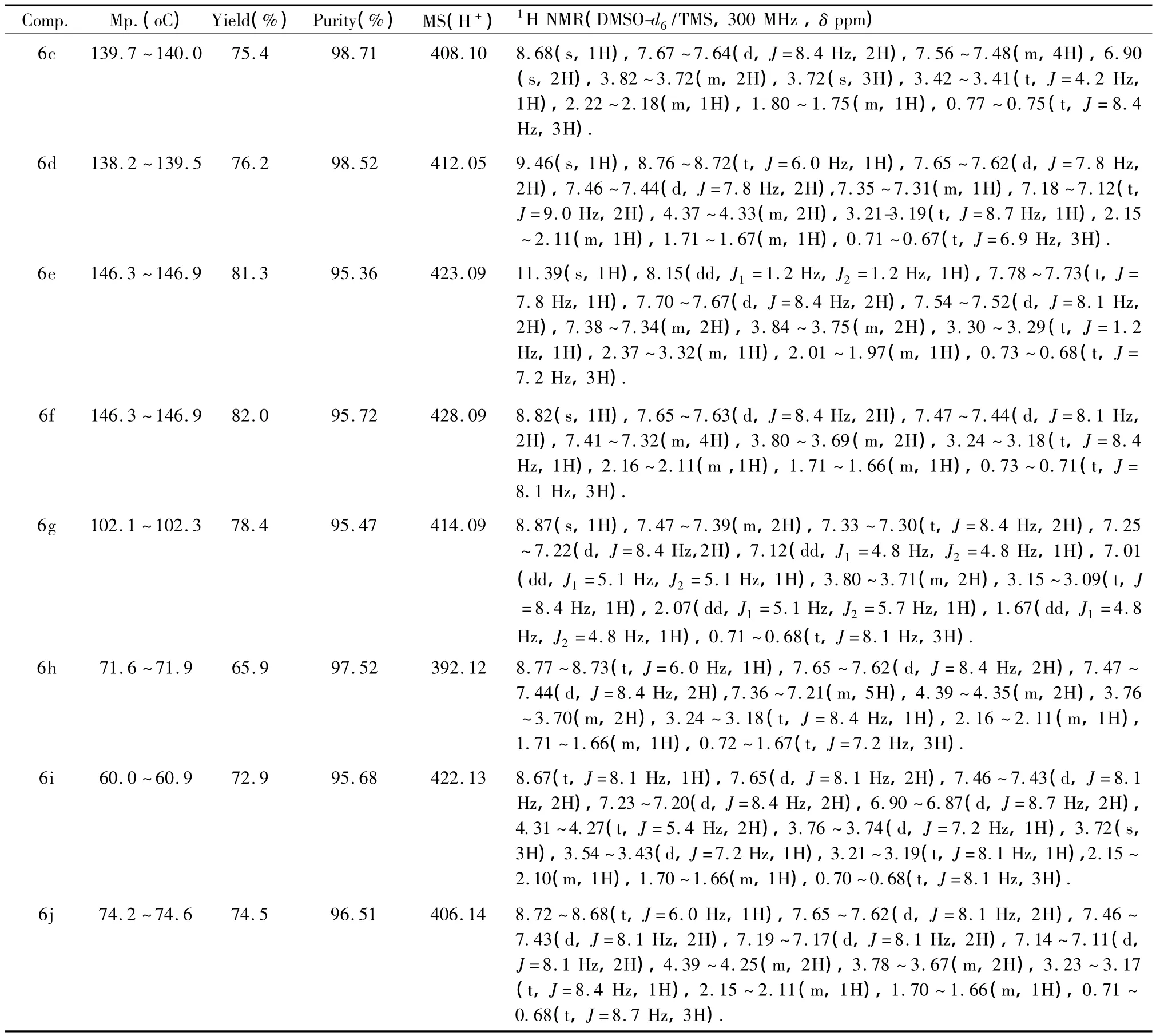
Comp. Mp.(oC) Yield(%) Purity(%) MS(H+) 1H NMR(DMSO-d6/TMS,300 MHz,δ ppm) 6c 139.7~140.0 75.4 98.71 408.10 8.68(s,1H),7.67~7.64(d,J=8.4 Hz,2H),7.56~7.48(m,4H),6.90 (s,2H),3.82~3.72(m,2H),3.72(s,3H),3.42~3.41(t,J=4.2 Hz,1H),2.22~2.18(m,1H),1.80~1.75(m,1H),0.77~0.75(t,J=8.4 Hz,3H). 6d 138.2~139.5 76.2 98.52 412.05 9.46(s,1H),8.76~8.72(t,J=6.0 Hz,1H),7.65~7.62(d,J=7.8 Hz,2H),7.46~7.44(d,J=7.8 Hz,2H),7.35~7.31(m,1H),7.18~7.12(t,J=9.0 Hz,2H),4.37~4.33(m,2H),3.21-3.19(t,J=8.7 Hz,1H),2.15~2.11(m,1H),1.71~1.67(m,1H),0.71~0.67(t,J=6.9 Hz,3H). 6e 146.3~146.9 81.3 95.36 423.09 11.39(s,1H),8.15(dd,J1=1.2 Hz,J2=1.2 Hz,1H),7.78~7.73(t,J= 7.8 Hz,1H),7.70~7.67(d,J=8.4 Hz,2H),7.54~7.52(d,J=8.1 Hz,2H),7.38~7.34(m,2H),3.84~3.75(m,2H),3.30~3.29(t,J=1.2 Hz,1H),2.37~3.32(m,1H),2.01~1.97(m,1H),0.73~0.68(t,J= 7.2 Hz,3H). 6f 146.3~146.9 82.0 95.72 428.09 8.82(s,1H),7.65~7.63(d,J=8.4 Hz,2H),7.47~7.44(d,J=8.1 Hz,2H),7.41~7.32(m,4H),3.80~3.69(m,2H),3.24~3.18(t,J=8.4 Hz,1H),2.16~2.11(m,1H),1.71~1.66(m,1H),0.73~0.71(t,J= 8.1 Hz,3H). 6g 102.1~102.3 78.4 95.47 414.09 8.87(s,1H),7.47~7.39(m,2H),7.33~7.30(t,J=8.4 Hz,2H),7.25~7.22(d,J=8.4 Hz,2H),7.12(dd,J1=4.8 Hz,J2=4.8 Hz,1H),7.01 (dd,J1=5.1 Hz,J2=5.1 Hz,1H),3.80~3.71(m,2H),3.15~3.09(t,J =8.4 Hz,1H),2.07(dd,J1=5.1 Hz,J2=5.7 Hz,1H),1.67(dd,J1=4.8 Hz,J2=4.8 Hz,1H),0.71~0.68(t,J=8.1 Hz,3H). 6h 71.6~71.9 65.9 97.52 392.12 8.77~8.73(t,J=6.0 Hz,1H),7.65~7.62(d,J=8.4 Hz,2H),7.47~7.44(d,J=8.4 Hz,2H),7.36~7.21(m,5H),4.39~4.35(m,2H),3.76~3.70(m,2H),3.24~3.18(t,J=8.4 Hz,1H),2.16~2.11(m,1H),1.71~1.66(m,1H),0.72~1.67(t,J=7.2 Hz,3H). 6i 60.0~60.9 72.9 95.68 422.13 8.67(t,J=8.1 Hz,1H),7.65(d,J=8.1 Hz,2H),7.46~7.43(d,J=8.1 Hz,2H),7.23~7.20(d,J=8.4 Hz,2H),6.90~6.87(d,J=8.7 Hz,2H),4.31~4.27(t,J=5.4 Hz,2H),3.76~3.74(d,J=7.2 Hz,1H),3.72(s,3H),3.54~3.43(d,J=7.2 Hz,1H),3.21~3.19(t,J=8.1 Hz,1H),2.15~2.10(m,1H),1.70~1.66(m,1H),0.70~0.68(t,J=8.1 Hz,3H). 6j 74.2~74.6 74.5 96.51 406.14 8.72~8.68(t,J=6.0 Hz,1H),7.65~7.62(d,J=8.1 Hz,2H),7.46~7.43(d,J=8.1 Hz,2H),7.19~7.17(d,J=8.1 Hz,2H),7.14~7.11(d,J=8.1 Hz,2H),4.39~4.25(m,2H),3.78~3.67(m,2H),3.23~3.17 (t,J=8.4 Hz,1H),2.15~2.11(m,1H),1.70~1.66(m,1H),0.71~0.68(t,J=8.7 Hz,3H).
2 药理实验
2.1 材料与方法 测试细胞株:肺癌细胞(A549),肝癌细胞(QGY),宫颈癌细胞(HeLa),结肠癌细胞(SW480)。阳性对照药:TG-10。测试方法:采用MTT法进行体外抗肿瘤活性筛选。
2.2 药理实验操作
2.2.1 细胞培养 受试细胞在5%CO2、37℃条件下,用含10%小牛血清的DMEM溶液传代培养,实验所用细胞均处于对数生长期。
2.2.2 药液制备 受试药物分别用DMSO配成10 g/ L溶液,-20℃保存,实验前,将药液取出置室温融化,并用10%小牛血清的DMEM培养液溶解分别成80、40、20、10、5、2.5 μg/ml的药液样品,4℃冰箱保存。
2.3 实验步骤
步骤1:取对数生长期细胞,用含10%小牛血清的DMEM培养液,制成单细胞悬液1×106个/ml,将该悬液加到96孔板中,每孔加入100 μl。
步骤2:在37℃培养箱中培养24 h后,吸取上清液,分别加入各浓度的受试药物,设双复孔,继续培养24 h。
步骤3:吸取上清液,加入20 μl MTT溶液(5 μg/ ml),继续培养4 h后,吸取上清液,加100 μl的DMSO,充分溶解后在570 nm处测定吸光值(OD值),并按下列公式计算其抑制率。
步骤4:计算IC50,抑制率(%)=(对照孔OD值-实验孔OD值)/对照孔OD值×100%,然后由SPSS软件算出IC50值。
2.4 抗肿瘤实验结果 见表2。
3 结果与讨论
采用微波反应,经knoevenagel condensation缩合、环化、水解及酰胺化等反应设计、合成了20个未见文献报道的反式构型的N-取代苯基-2-(4-取代苯基)环丙烷-1-甲酸乙酯-1-酰胺系列化合物,化合物的结构经1H NMR和LC-MS确证。合成中利用微波反应,比文献报道[8]的合成方法在保持收率情况下,节省了原材料,降低了环境污染及提高了合成效率。体外抗肿瘤活性筛选实验结果显示:20个目标化合物对4种肿瘤细胞具有一定的抑制活性。其中,对A549非小细胞肺癌的抗肿瘤活性最好。对氯苯基取代系列化合物(5a~5j)比对三氟甲基苯基取代系列化合物(6a~6j)有更好的抗肿瘤活性(5h对A549、HeLa、QGY肿瘤细胞的IC50分别为18.3、24.6、56.2 μM,而6h相对应的肿瘤细胞活性IC50分别为46.2、36.1 μM和大于400 μM;苄胺取代比苯胺取代的活性较优,5i对4种肿瘤细胞株的IC50分别为34.2、38.1、49.6、56.4 μM,相对比5c的IC50分别为53.5、89.9、167.5、90.5 μM;在酰胺芳香环上的不同取代基对抗肿瘤活性体现出一定的规律性:吸电子基团优于供电子基团,化合物5b的化合物活性比5a活性较高,6b的化合物活性比6a较高。综合所有的化合物抗肿瘤活性结果,化合物5b具有最好的活性,对A549、HeLa细胞的IC50分别达到了6.8、12.40 μM,优于阳性对照药TG-10(12.8、18.3 μM)。

表2 目标化合物体外抗肿瘤活性(IC50,μM)
笔者对TG-10进行了结构修饰,合成了20个全新的环丙烷酰胺类化合物,并进行了体外抗肿瘤活性筛选。肿瘤活性筛选结果表明,该类化合物具有一定的抗肿瘤活性,并具有一定的药物构效关系。其中,化合物5b对A549、HeLa的活性优于TG-10,取得了较好的效果。但由于所合成的化合物数量有限,尚未对该类化合物药效关系进行更全面的研究。因此,有待我们对该类化合物进行更深层次的研究,建立环丙烷酰胺类化合物的数据库,为该类化合物研究奠定基础。
[1] 王甜甜,李 科.四氢呋喃木脂素、四氢呋喃并[3,4-c]吡喃-4-酮、2,5-二氢呋喃以及呋喃衍生物的设计、合成和抗肿瘤活性研究[D].第二军医大学博士论文,2011.
[2] 姜殿君,赵丽妮,崔 晶.抗肿瘤药物治疗的研究进展[J].中国现代医药杂志,2009,11(4):8.
[3] 周大铮,易杨华,毛士龙.香榧假种皮中的木脂素成分[J].药学学报,2004,39(4):269.
[4] 周大铮,易杨华.香榧中抗艾滋病病毒活性先导化合物的研究和结构修饰[D].第二军医大学博士论文,2004.
[5] Wang TT,Liu J,Li K,et al.Efficient and mild synthesis of highly substituted 2,5-dihydrofuran and furan derivatves via stepwise reaction[J].Tetrahedron,2011,67(7):3476.
[6] Sun HL,Wang TT,Li K,et al.Synthesis,chiral resolution,and determination of novel furan lignan derivatives with potent antitumor activity[J].Bioorg Med Chem Lett,2010,20(11):1961.
[7] Wang TT,Liu J,Li K,et al.Synthesis and anti-tumor activity of novel ethyl 3-aryl-4-oxo-3,3a,4,6-tetrahydro-1H-furo[3,4-c]pyran-3a-carboxylates[J].Bioorg Med Chem Lett,2011,21 (9):3381.
[8] He XR,Qiu GP,Yang J,et al.Synthesis and anticonvulsant activity of new 6-methyl-1-substituted-4,6-diazaspiro[2.4]heptane-5,7-diones[J].Eur J Med Chem,2010,45(6):3818.
