变异链球菌核糖-5-磷酸异构酶A的表达、纯化与鉴定
2012-01-25武文琦丛旭珍殷爱红翟方丽李慎涛
武文琦 丛旭珍 殷爱红 胡 家 翟方丽 李慎涛*
(1.首都医科大学基础医学院生物化学与分子生物学和免疫学系,北京100069;2.首都医科大学医学实验与测试中心,北京100069;3.青岛阜外心血管病医院检验科,青岛266034)
变异链球菌(streptococcus mutans)是龋病的主要致病菌,也是某些非口腔疾病(如亚急性细菌性心内膜炎)的病原菌,具有广泛的适应能力,可以持续存在于口腔中。在世界范围内,不同种族、不同社会经济背景的人群中均可分离出此菌,但并非所有人都表现龋病。研究[1]显示,不同龋指数患者口腔变异链球菌的体外致龋能力不同,提示不同菌株的致龋性是不同的,而致龋性主要是通过其毒力因子来完成的,但其感染途径和致病机制至今尚未得到满意的解答。随着S.mutans UA159基因组测序工作的完成,相关的理论研究[4]也更加深入细致。
核糖-5-磷酸异构酶也叫D-核糖-5-磷酸乙酮醇异构酶,是自然界中广泛存在的一种高度保守的蛋白酶,参与原核和真核生物的糖代谢(如磷酸戊糖途径),同时在植物二氧化碳固定的卡尔文循环中,它是催化5-磷酸核糖和5-磷酸核酮糖相互转变的关键酶[2-3]。这些代谢参与还原型辅酶Ⅱ(NADPH)的合成以及戊糖的氧化、非氧化合成[4]。有研究[5]表明,人类缺乏核糖-5-磷酸异构酶会引起髓鞘的损伤,从而导致诸如白质脑病以及包括周围神经病变等在内的神经元异常。核糖-5-磷酸异构酶以核糖-5-磷酸异构酶A(RpiA)和核糖-5-磷酸异构酶B(RpiB)这2种类型存在,它们都能催化5-磷酸核糖和5-磷酸核酮糖的相互转变,但是它们却几乎没有什么关联。RpiA是自然界中最广泛存在的,几乎存在于所有的组织中[6],而 RpiB却只在细菌和真核物种中发现[7]。RpiA和RpiB能够催化相同的反应并且在大部分的器官中至少存在一种。尽管核糖-5-磷酸异构酶的重要性早已被确定,但对RpiA和RpiB的催化机制却所知甚少。
本文中所研究的rpiA由变异链球菌(streptococcus mutans,Smu)UA159株基因组所编码,相对分子约为24 000。此菌属革兰阳性菌,是最主要的龋齿致病菌之一。对rpiA克隆、表达与纯化将有助于阐明其生物学功能,通过寻找其相互作用蛋白及小分子化合物,为进一步研制抗变异链球菌的药物提供理论基础。
1 材料和方法
1.1 材料和试剂
Streptococcus mutans UA159由北京大学口腔医学院郭丽红教授惠赠;原核表达载体pGEX-6p-1、大肠杆菌克隆菌株XL-1及表达菌株BL21(DE3)均为本实验室保存;DNA聚合酶、各种限制性内切酶和T4 DNA连接酶购自NEB公司(美国);DNA片段胶回收试剂盒、质粒提取试剂盒购自美国Omiga公司;Glutathione SepharoseTM4B购自美国 GE公司;Sephadex G25、Resource Q预装柱购自美国GE公司。引物合成及DNA测序由上海生工公司完成。
制冷恒温摇床,Thermo公司;超声波细胞粉碎机,宁波新芝生物科技股份有限公司JY92-2D;电泳仪,BIO-RAD,PowerPac3000;高速离心机,BECKMAN J2-HS;凝胶成像处理系统,GE healthcare Image Quant 300;AKTA purifier蛋白纯化系统,美国 GE公司; MALDI-TOF-TOF质谱仪,德国BRUKER公司。
1.2 方法
1)目的基因片段的扩增:根据GenBank中变异链球菌UA159株基因组(AE014133)序列,设计特异性引物,分别添加BamHI和XhoI酶切位点。以UA159基因组为模板,PCR扩增核糖-5-磷酸异构酶A的编码序列。将PCR产物进行1%琼脂糖凝胶电泳,用DNA回收试剂盒回收PCR产物。
2)克隆及鉴定:将胶回收的PCR产物用BamHI和XhoI双酶切,回收酶切后的片段,与经过同样双酶切的载体pGEX-6p-1按5∶1浓度混合,在T4 DNA连接酶作用下于16℃连接过夜,用连接产物转化E coli XL-1 blue,涂布到含氨苄青霉素(100 mg/L)的LB平板上,于37℃培养过夜,次日从LB平板上采用抽签法随机挑取5个菌落,接种于含氨苄青霉素(100 mg/ L)的LB培养基中,于37℃培养16 h,提取质粒,进行PCR鉴定,将PCR鉴定为阳性的质粒再进行酶切鉴定,将酶切鉴定为阳性的质粒测序。
3)诱导表达分析:用测序正确的质粒转化大肠杆菌BL21(DE3),采用抽签法随机挑取5个菌落,接种于含氨苄青霉素(100 mg/L)的LB培养基中,于37℃培养过夜,次日,按1∶100的比例将过夜培养物转接到5 mL含氨苄青霉素(100 mg/L)的LB液体培养基中,于37℃培养,当培养液OD600值达到0.6~0.8时,加入IPTG(终浓度1 mmol/L),诱导表达8~12 h,5 000 r/min离心15 min收集菌体,取适量菌体用 SDSPAGE分析蛋白表达情况。
4)蛋白表达形式分析及表达条件的优化:分别对在不同诱导温度、不同IPTG终浓度、不同诱导起始培养液OD600值及不同诱导时间4种条件下诱导蛋白表达,4 000 r/min离心15 min收集菌体,用PBS(140 mmol/L NaCl、2.7 mmol/L KCl、10 mmol/L Na2HPO4·12H2O、1.8 mmol/L KH2PO4)重悬菌体,4 000 r/min离心15 min,收集菌体,用PBS重悬菌体,加入溶菌酶(终浓度至1 g/L)、DTT(终浓度至1 mmol/ L)、PMSF(终浓度至1 mmol/L),冰上作用30 min,然后,在冰浴上超声至菌体完全裂解,15 000 r/min离心50 min,分别收集上清和沉淀,用SDS-PAGE分上清和沉淀中的目标蛋白,从而确定目标蛋白的表达形式。
5)重组蛋白的诱导表达:将核糖-5-磷酸异构酶A工程菌接种于20 mL含氨苄青霉素(100 mg/L)的LB液体培养基中,37℃振荡培养过夜,次日按1∶100的体积比转接于1 L含氨苄青霉素(100 mg/L)的LB液体培养基中,于37℃培养,当培养液OD600值达到0.6时,加入IPTG(终浓度0.1 mmol/L),于16℃下诱导表达20 h,5 000 r/min离心15 min收集菌体用于目标蛋白的纯化。
6)重组蛋白的纯化:将诱导表达的菌体用超声破菌,离心收集上清,加到已用PBS平衡的Glutathione SepharoseTM4B亲和层析柱上,让其自然通过柱,然后用PBS洗涤杂蛋白,用酶切缓冲液(25 mmol/L Tris、200 mmol/L NaCl、2 mmol/LDTT,pH 7.5)平衡柱,加入200μL(1 g/L)PreScission蛋白酶,于4℃酶切过夜。次日,用 PBS洗脱,收集穿过的蛋白样品,用15%的SDS-PAGE鉴定。
收集目标蛋白,用Sepherdex G25柱脱盐,缓冲液用阴离子交换A液(25 mmol/L Tris、1 mmol/L DTT,pH 7.5),随后将脱盐后的蛋白样品过Resource Q阴离子交换柱,所用缓冲液为:A液(25 mmol/L Tris、1 mmol/L DTT,pH 7.5)、B液(25 mmol/L Tris、1 mol/L NaCl、1 mmol/L DTT,pH 7.5)。收集含蛋白组分,用SDS-PAGE鉴定,将含有目标蛋白的组分合并,利用AKTA纯化仪随机软件对洗脱峰进行积分并估算蛋白质浓度。
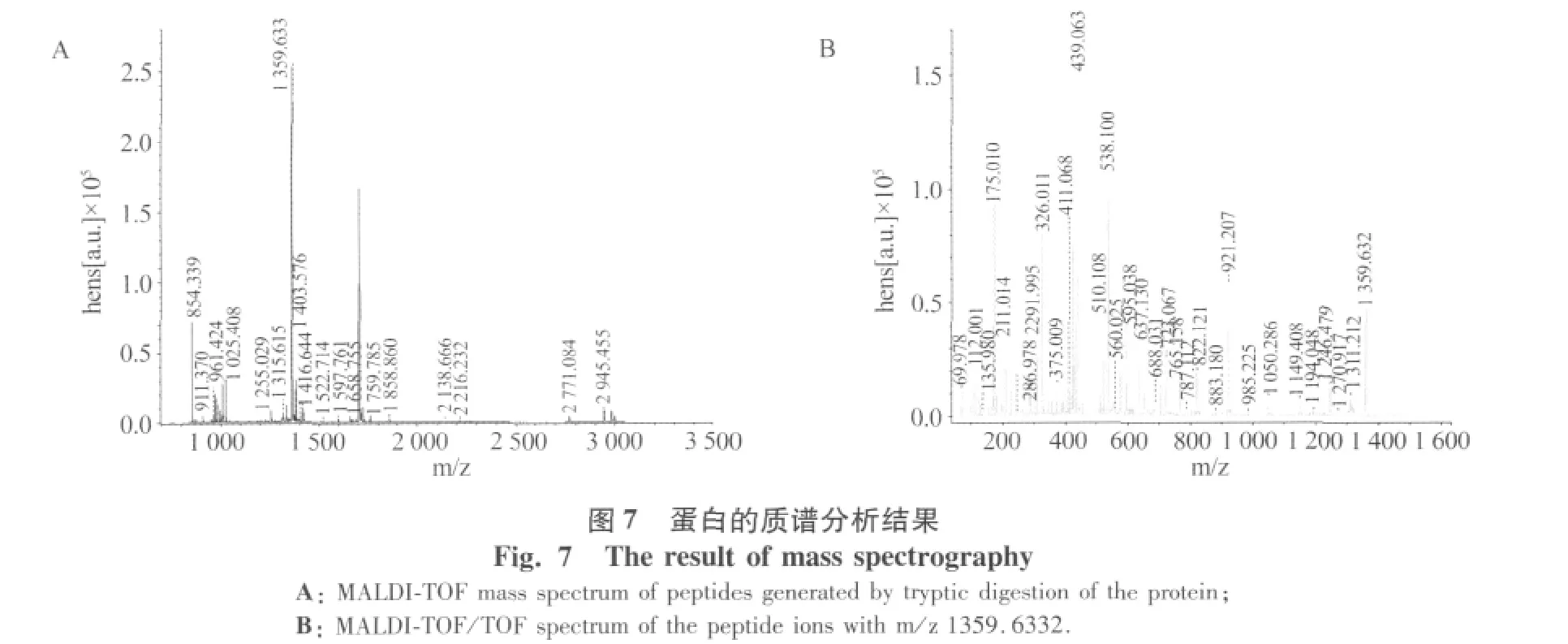
7)目标蛋白的质谱分析:从SDS-PAGE胶上切取含有目标蛋白质的条带,经胶内酶解后,用MALDITOF-TOF质谱仪进行分析,运用Mascot软件将质谱结果在NCBInr数据库中进行检索,得出蛋白质的可能性分数,根据所用蛋白数据库来确定阳性的分数,从而判定是否为核糖-5-磷酸异构酶A蛋白。
2 结果
2.1 RpiA表达载体的构建
PCR扩增得到了编码rpiA的DNA片段,经1%琼脂糖凝胶电泳鉴定,大小与预期的一致。将该片段和pGEX-6p-1载体连接后,转化大肠杆菌XL-1 blue,挑取单菌落,经菌液PCR鉴定得到阳性克隆,经质粒酶切鉴定,得到阳性克隆,测序结果证实为核糖-5-磷酸异构酶A蛋白编码序列。
2.2 诱导表达及蛋白表达形式分析
将rpiA工程菌于37℃、1 mmol/L IPTG的条件下诱导表达20 h,超声破碎菌体,4℃、1 5000 r/min离心30 min,分别收集上清和沉淀,进行SDS-PAG分析。在破菌沉淀中可见相对分子质量约为40 000的融合蛋白表达条带,大小与预期的一致,上清中也含有少量目标蛋白,说明rpiA能够在大肠杆菌中高效表达,但在37℃诱导条件下,表达产物主要以包含体的形式存在。
2.3 蛋白表达条件的优化
1)在培养液 OD600值等于0.8、IPTG浓度0.1 mmol/L条件下,分别在37℃、16℃诱导菌体表达目标蛋白,于诱导后取破菌上清进行SDS-PAGE,结果表明16℃时目标蛋白在上清中的表达量较大(图1)。

图1 目标蛋白在不同温度下诱导后的SDS-PAGEFig.1 SDS-PAGE analysis of target protein induced with IPTG at different temperature1:before induction;2:after induction at 16℃;3:16℃ supernatant;4:before induction;5:after induction at 37℃;6:37℃supernatant;M:66 000,46 000,34 000,26 000,16 000; IPTG:isopropyl β-D-1-thiogalactopyranoside;M:marker.
2)在培养液OD600值等于0.8、16℃条件下,用终浓度分别为0.1、0.2、0.3、0.4、0.5、0.6、0.7、0.8 mmol/L的IPTG诱导菌体表达目标蛋白,于诱导后20 h取样SDS-PAGE,结果表明当IPTG终浓度为0.5 mmol/L、4 mmol/L时目标蛋白的表达量最大(图2)。
3)在培养液OD600值等于0.8、16℃、IPTG终浓度为0.5 mmol/L条件下诱导菌体表达目标蛋白,于诱导后1 h、3 h、5 h、7 h、9 h、11 h、20 h取样SDSPAGE,结果表明诱导20 h目标蛋白的表达量最大(图3)。
4)在IPTG终浓度为0.1 mmol/L、16℃诱导条件下,在培养液OD600值分别为0.4、0.6、0.8、1.0、1.2时诱导菌体表达目标蛋白,于诱导后20 h取样SDS-PAGE,结果表明当培养液OD600值为0.6时开始诱导目标蛋白的表达量最大(图4)。



由上述实验可知,变异链球菌核糖-5-磷酸异构酶A在大肠杆菌中的最佳表达条件为:培养温度16℃、开始诱导的培养液OD600值等于0.6、IPTG终浓度0.5 mmol/L、诱导时间20 h。
2.4 蛋白纯化
将收集的菌体破碎、离心后,收集上清,进行Glutathione SepharoseTM4B亲和层析纯化,得到较高纯度的目的蛋白。合并后经电泳鉴定,得到较高纯度的目的蛋白(图5)。
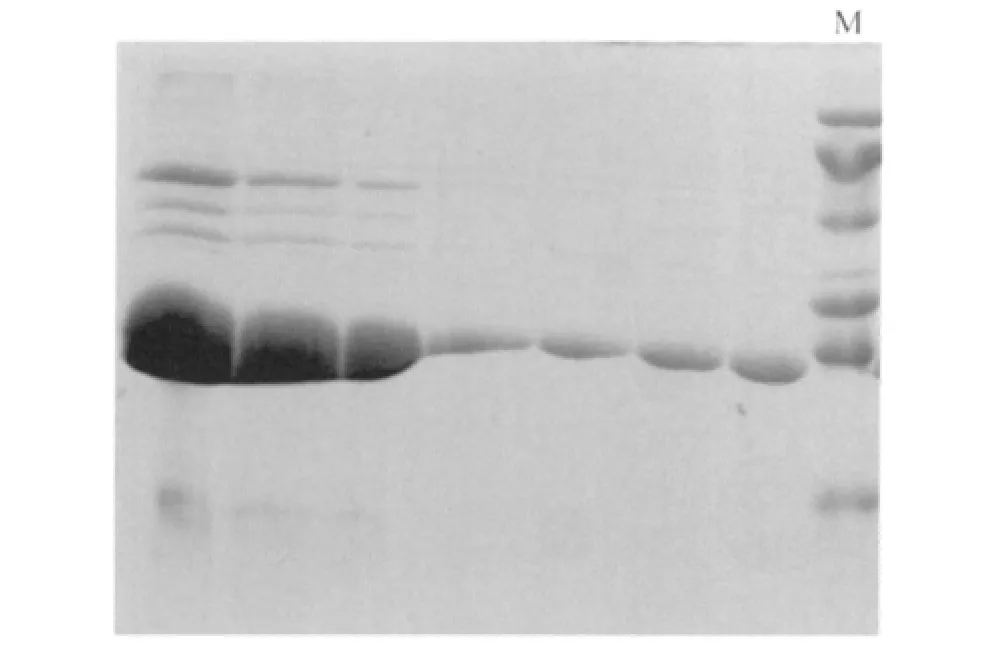
图5 Glutathione SepharoseTM4B亲和层析纯化后目标蛋白的SDS-PAGEFig.5 SDS-PAGE analysis of rpiA eluted from affinity chromatographyM:marker.
利用阴离子交换进一步纯化目的蛋白,收集主峰各组分,合并后经电泳鉴定,得到较高纯度的目的蛋白(图6)。
2.5 蛋白的质谱鉴定
将SDS-PAGE凝胶上的样品条带切下进行胶内酶切,再进行MALDI-TOF-TOF质谱分析,经数据库搜索后,结果为变异链球菌核糖-5-磷酸异构酶A,得分178,是可信的匹配结果。因此从蛋白水平证实该蛋白为变异链球菌核糖-5-磷酸异构酶A(图7)。
3 讨论
在磷酸戊糖途径中,核糖-5-磷酸和核酮糖-5-磷酸之间的相互转化是至关重要的一步。磷酸糖异构酶能够参与催化非磷酸化的醛糖或者酮糖之间的相互转化,尽管催化效率不如它们自然的磷酸化底物高,至少在核糖-5-磷酸异构酶中是如此[8]。由于部分糖类异构酶具有广泛的底物特异性,许多酶比如6-磷酸半乳糖苷酶已经用于罕见糖类的异构化[9]。这些异构酶通过2种不同的异构化反应将一种底物转化成2种不同的产物。与这些酶的异构化反应所不同的是,核糖-5-磷酸异构酶催化糖类的相互转变则只是通过一种异构化反应。RpiA和RpiB这2种完全没有氨基酸序列相似性的酶的催化活性首先在大肠杆菌中被确证,在研究过程中,RpiA被认为是促进生命活动过程中所必须的五碳糖之间的相互转变,而RpiB的主要作用则是促进罕见六碳糖类6-磷酸核糖和6-磷酸核酮糖的相互转化。对于变异链球菌RpiA的异构化作用及其对于底物的相对特异性选择则需要通过相关的动力学特征研究来得到进一步的确证。

RpiA已经从至少60个物种中获得序列并且已经在许多有机体中得到了克隆,包括大肠杆菌和菠菜。这些酶之间以及与人类体内的酶之间的氨基酸序列均具有30%的同源性。细菌rpiA参与细菌的磷酸戊糖途径,对细菌的生存至关重要。变异链球菌是重要的致龋菌,但目前缺乏有针对性的治疗药物,对其致龋机制也缺乏深入的了解。寻找新的分子靶标,特别是与其毒力相关的蛋白质成为目前研究的热点。通过对这类蛋白功能的进一步研究,有望在分子水平上理解变异链球菌的致龋机制,对开发有针对性的治疗药物或疫苗有极大帮助。
本研究成功地在大肠杆菌中表达了变异链球菌rpiA,并建立了高效纯化工艺。目前正在进行rpiA结构与功能方面的研究,探索变异链球菌rpiA在致龋方面的作用机制。
[1] Ajdic D,McShan W M,McLaughlin R E,et al.Genome sequence of Streptococcus mutansUA159,a cariogenic dental pathogen[J].Proc Natl Acad Sci U S A,2002,99(22):14434-14439.
[2] Wamelink M M,Struys E A,Jakobs C.The biochemistry and inherited defects of the pentose phosphate pathway:a review[J].J Inherit Metab Dis,2008,31(6):703-717.
[3] Roos A K,Mariano S,Kowalinslai E,et al.D-ribose-5-phosphate isomerase B from Escherichia coli is also a functional D-allose-6-phosphate ismerase,while the Mycobacterium tuberculosis enzyme is not[J].J Mol Biol,2008,382 (3):667-679.
[4] Melendez-Hevia E,Waddell T G,Heinrich R,et al.Theoretical approaches to the evolutionary optimization of glycolysis-chemical analysis[J].Eur J Biochem,1997,244(2): 527-543.
[5] Huck J H,Verhoeven N M,Struys E A,et al..Ribose-5-phosphate isomerase deficiency:new inborn error in the pentose phosphate pathway associated with a slowly progrssive leukoencephalopathy[J].Am J Hum Genet,2004,74(4):745-751.
[6] Ishikawa K,Matsui I,Payan F,et al.A hyperthermostable D-ribose-5-phosphate isomerase from Pyrococcus horikoshii characterization and three-dimensional structure[J].Structure(Camb),2002,10(6):877-886.
[7] Zhang R G,Andersson C E,Skarina T,et al.The 2.2 resolution structure of RpiB/AlsB from Escherichia coli illustrates a new approach to the ribose-5-phosphate isomerase reaction[J].J Mol Biol,2003,332(5):1083-1094.
[8] Yeom S J,Kim B N,Park C S,et al.Substrate specificity of ribose-5-phosphate isomerases from Clostridium difficile and Thermotoga maritima[J].Biotechnol Lett,2010,32 (6):829-835.
[9] Park H Y,Park C S,Kim H J,et al.Substrate specificity of a galactose 6-phosphate isomerase from Lactococcus lactis that produces D-allose from Dpsicose[J].J Biotechnol,2007,132(1):88-95.
