Sequence analysis of envelope genes in dengue viruses from Fujian Province,2004-2010
2012-01-24HUANGMengZHANGYongjunLINMeiqingWANGJinzhangYANYanshengWENGYuwei
HUANG Meng,ZHANG Yong-jun,LIN Mei-qing,WANG Jin-zhang,YAN Yan-sheng,,WENG Yu-wei
(1.Fujian Priority Laboratory for Zoonoses,Fujian Center for Disease Control and Prevention,Fuzhou350001,China;2.Teaching Base for School of Public Health,Fujian Medical University,Fuzhou350001,China;3.School of Public Health,Fujian Medical University,Fuzhou350108,China)
Dengue fever(DF),an anthrop-borne viral disease that mainly circulates in tropical and sub-tropical regions of the world,is caused by dengue viruses(DENV)of 4serotypes.It was estimated by the World Health Organization(WHO)that approximately more than 40%of the world's population is at risk of dengue thus far,accounting for nearly 50-100million new dengue infections worldwide every year.Moreover,among the annual 500 000 cases with severe manifestation including dengue hemorrhagic fever(DHF)or dengue shock syndrome(DSS),about 2.5%of them are fatal[1].During the past 5decades,the gradually expansion of DF has become a global public health problem.
As a coastal province in Southeast China,Fujian has expanded its frequent exchange with foreign countries and regions in trade,tourism and cultural activities,etc.The number of entry-and exit-port passengers in Fujian is steadily increasing recent years,while certain infectious diseases including DF has been introduced to Fujian and occasionally spread in local residents.Imported DF cases have been reported in Fujian almost every year since 1999,even contributed to some local outbreaks[2-3].Thus,aprovince-wide DF surveillance network including etiology surveillance was launched in Fujian from 2005,which was an important control and prevention measure in the past seven years in tracking the ongoing situation of DF epidemic and implementing the timely disease control means.In this study,viral envelope(E)genes of DENV strains from Fujian in recent years were sequenced and analyzed,and the possible origins of these strains were deduced in combination with epidemiology investigation data during2004 -2010.These results would provide virological evidence for DF surveillance in the near future,highlighting current priorities of DF control and prevention in Fujian.
Materials and Methods
DENV strains in this study were isolated from serum specimens in confirmed DF patients from outbreaks or sporadic cases during2004 -2010.Virus isolation was conducted at Department of Viral Disease Control,Fujian Center for Disease Control and Prevention(Table 1).Specimens were in-oculated on C6/36cells.Each strain was identified by immunofluorescence or RT-PCR[4],and stored in a freezer at-80℃.Viral RNA was extracted from 140μL of virus stock using QIAamp Viral RNA Mini Kit(QIAGEN,Germany),and finally eluted in 50μL DEPC-treated H2O and stored at-20℃until further use.
Viruses and RNA extraction
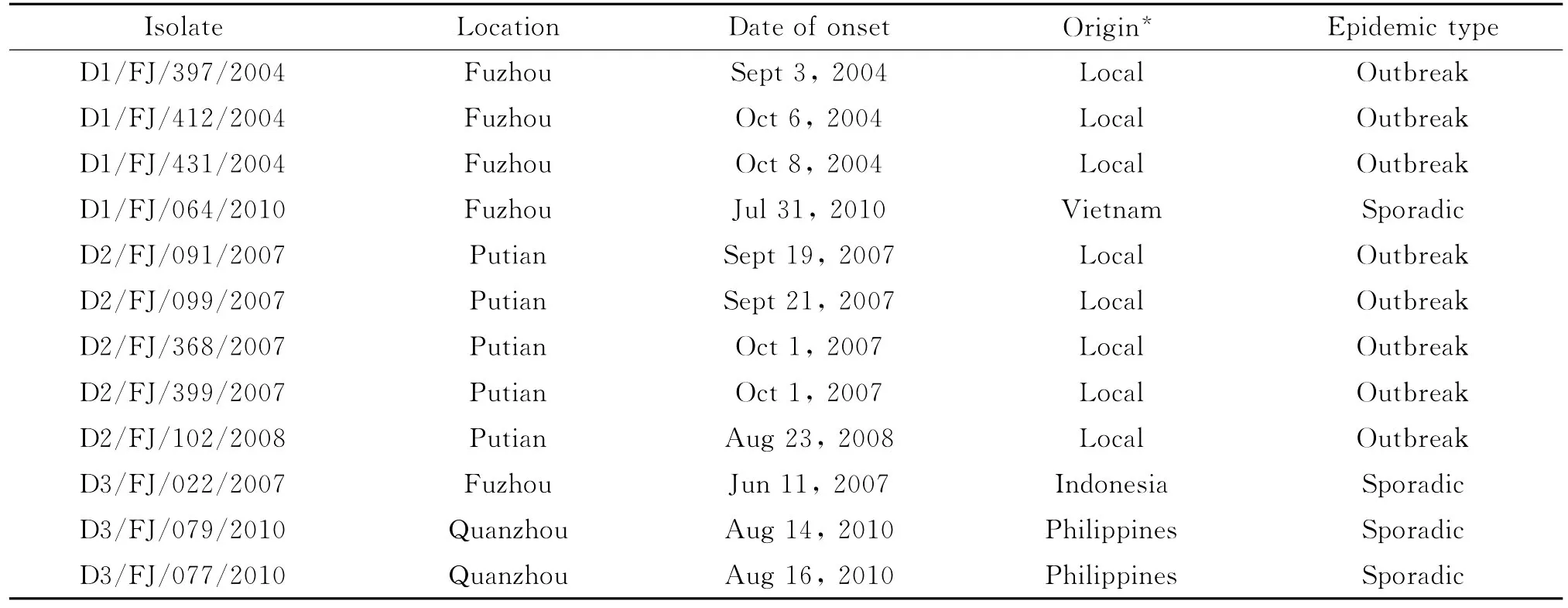
Isolate Location Date of onset Origin* Epidemic type D1/FJ/397/2004 Fuzhou Sept 3,oradic 2004 Local Outbreak D1/FJ/412/2004 Fuzhou Oct 6,2004 Local Outbreak D1/FJ/431/2004 Fuzhou Oct 8,2004 Local Outbreak D1/FJ/064/2010 Fuzhou Jul 31,2010 Vietnam Sporadic D2/FJ/091/2007 Putian Sept 19,2007 Local Outbreak D2/FJ/099/2007 Putian Sept 21,2007 Local Outbreak D2/FJ/368/2007 Putian Oct 1,2007 Local Outbreak D2/FJ/399/2007 Putian Oct 1,2007 Local Outbreak D2/FJ/102/2008 Putian Aug 23,2008 Local Outbreak D3/FJ/022/2007 Fuzhou Jun 11,2007 Indonesia Sporadic D3/FJ/079/2010 Quanzhou Aug 14,2010 Philippines Sporadic D3/FJ/077/2010 Quanzhou Aug 16,2010 Philippines Sp
Note:* means infection location was deduced according to the case investigation.
Primer design and RT-PCR
DENV genome sequences of reference strains available in GenBank database were downloaded.Primers for RT-PCR were designed using Vector NTI v11.0software and synthesized by Sangon Biotech(Shanghai)Co.,Ltd(Table 2).For amplification of full-length E genes,5μL of RNA template was added into 20μL of RT-PCR mixture containing 5μL 5× Buffer,1μL of dNTPs(10mmol/L each),1μL of Enzyme mix,1.5μL of forward and reverse primer(10μmol/L),and 10μL of DEPC-treated H2O,respectively.Reverse transcription was performed at 50℃for 30min,95℃for 15min,following by 35cycles of amplification consisting of 94℃for 30sec,annealing at 50℃for 30sec,and 72℃for 90sec,with a final extension at 72℃for 10min.Finally,RTPCR products were analyzed on 1%agarose gel.
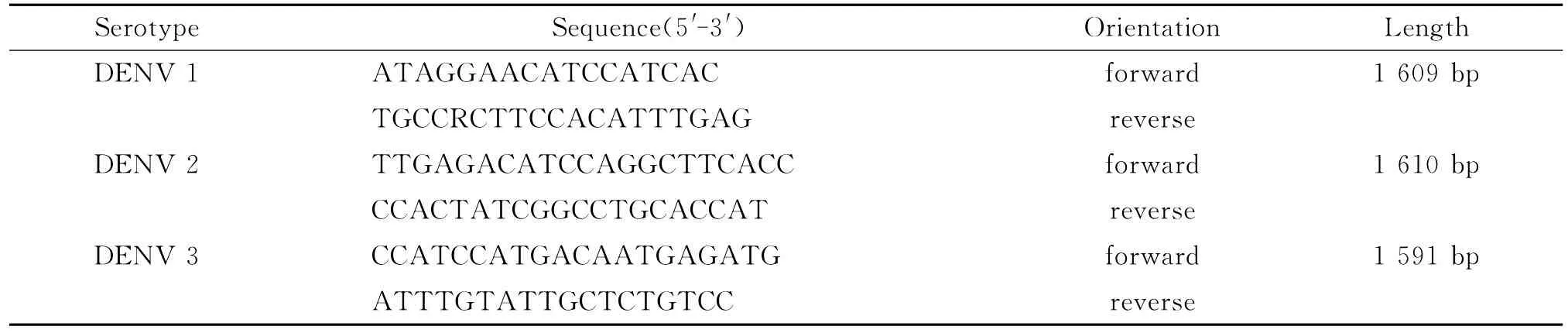
Tab.2 Primer sequences used in RT-PCR
TA cloning and plasmid DNA preparation
DNA fragments were gel purified by using QIAquick Gel Extraction Kit(QIAGEN,Germany),and cloned into pMD 19-T vector(TaKaRa,China).The recombinant plasmid was transformed into E.coli DH5αcompetent cells(TaKaRa,China),and selected on amp+ LB agar plates containing IPTG and X-gal overnight at 37℃.White colonies on LB agar plates were inoculated in amp+LB media,and cultured overnight on a shaker at 37℃.Plasmid DNA of 1mL cultured LB media was extracted by TIANprep Mini Plasmid Kit(TIANGEN,China),subsequently sequenced with RV-M and M13-47primers by TaKaRa Biotechnology Co.,LTD(Dalian,China).
Evolutionary analysis
Selected genome sequences of DENV strains during2004 -2010were obtained from GenBank database as reference sequences using inquiry sub-jects"dengue virus X complete genome"(X refers to 1,2,3,respectively).Full-length E genes of Fujian isolates and reference strains were aligned using MEGA v5.0software.Phylogenetic trees were constructed by Bayesian analysis(MrBayes v3.2),which is more accurate than Maximum Likelihood(ML)and Maximum Parsimony(MP)methods[5].Incorporating with the GTR+ Γ +I model of evolution,a Markov chain-Monte Carlo method(MCMC)was used to calculate posterior probabilities.The chain was sampled every 100th generation,until the average standard deviation of split frequencies was less than 0.01.After discarding the first 25%samples from cold chain,phylogenetic trees based on calculated branch lengths were reconstructed,and then generated by Tree-View program.
Results
Predicted amplicons of E genes were obtained from all 12DENV isolates from Fujian.It was indicated from sequencing data that E genes of both DENV-1and DENV-2were 1 485bp in length,encoding 495amino acids;E gene in DENV-3was 1 479bp encoding 493amino acids.No insertion or deletion was observed.BLAST analyses also showed that strains sharing the maximum identity sequences with the 12isolates originated from Southeast Asia(Table 3).

Tab.3 BLAST analyses for DENV E genes
Phylogenetic trees for each serotype of DENV were reconstructed,as shown in Figure 1,where the consensus tree for DENV-1was obtained by virtual computation for 100 000generations,DENV-2for 200 000,and DENV-3for 120 000 generations,respectively.Since the branch length in the trees represented its relative evolutionary distance,it was revealed that E genes of these 12 DENV isolates from Fujian were,in each serotype,highly homologous with those strains circulating in Southeast Asia,despite of longer genetic distance with isolates from other regions of the world.
Discussion
DENV,with a genome of single positive stranded RNA,belongs to the family Flaviviridae,genus Flavivirus.Like other RNA viruses,DENV is prone to mutate during viral replication for lack of accurate correction mechanisms.Consequently,the variant virus is likely to cause regional DF epidemic[6].Therefore,identification of virus mutation will contribute to determine transmission origins,describe etiological characterization and prevalence of DENV,and track introduction sources in DF outbreaks.E protein is one of the main structural proteins of DENV particles,and can mediate attachment of virions to host cell receptors and virus entry into host cells by membrane fusion.Stimulation of multiple epitopes in the E protein also induces neutralizing antibodies in immune systems of the host[7].Thus,E gene was widely considered as an important target gene in virus genotyping,evolution and transmission dynamics.In this study,full-length E gene sequences of DENV were analyzed to determine possible origins of DENV strains in Fujian Province,2004-2010.

Fig.1 Phylogenetic tree for E genes of DENVStrains are denoted by country of isolation,year of isolation and GenBank accession number.*:Epidemic strains in Southeast Asia
It was indicated from BLAST search and phylogenetic analyses that DENV strains in Fujian Province during2004 -2010were most similar to and highly homologous with those isolates circulating in Southeast Asia,confirming previous conclusions based on epidemiological investigations,suggesting that DF cases in Fujian were mainly introduced from Southeast Asia countries,occasionally causing local DF epidemic[8].So far,DENV has widely spread to more than 70countries globally.However,there is no evidence supporting that DF is endemic in China.Therefore,the most effective strategy for DF prevention and control in China is to monitor the introduction of DENV at entryports,i.e.,implementing extensive surveillance of passengers from DF endemic regions.These results suggested that inspection and quarantine should be strengthened for passengers from Southeast Asia at entry ports of Fujian.
It was revealed from full-length sequencing and E gene sequences analyses that three DENV-1 strains from a DF outbreak in Fuzhou,2004,were highly homologous[2].Another four DENV-2isolates from the 2007DF outbreak in Hanjiang Dis-trict,Putian,also showed identical genetic characterization[3].Moreover,two DENV-3strains(D3/FJ/079/2010and D3/FJ/077/2010)isolated in returned tourists after a trip to Philippine in 2010 shared high homology.Such cases were all introduced by a single transmission source,which consistent with conclusions from epidemiological investigation.Notably,considerable genetic difference was observed between DENV-2strains in 2007and that in 2008from Hanjiang,Putian,demonstrating that the DF case in 2008was caused by another introduction from Southeast Asia,rather than localized transmission of the 2007outbreak.In summary,sequencing and phylogenetic analysis of E genes in DENV isolates contribute to tracking transmission origins,and support investigation of epidemiological association among cases during DF outbreaks.
[1]WHO.Dengue and severe dengue[EB/OL].http://www.who.int/mediacentre/factsheets/fs117/en/,2012-01.
[2]Yan YS,Hong RT,Shen XN,et al.Epidemiological and etiological characterization of a dengue fever outbreak in Fuzhou,2004[J].Chin J Epidemiol,2006,27(5):371-374.(in Chinese)严延生,洪荣涛,沈晓娜,等.福州市2004年登革热流行病学和病原学特征分析[J].中华流行病学杂志,2006,27(5):371-374.
[3]Weng YW,Hong RT,Zhang SY,et al.Epidemiological investigation of a dengue fever outbreak in Putian,Fujian province,2007[J].Chin J Zoonoses,2009,25(4):330-333.(in Chinese)翁育伟,洪荣涛,张山鹰,等.福建省莆田市2007年登革热暴发的流行病学调查分析[J].中国人兽共患病学报,2009,25(4):330-333.
[4]Lanciotti RS,Calisher CH,Gubler DJ,et al.Rapid detection and typing of Dengue viruses from clinical samples by using reverse transcriptase-polymerase chain reaction[J].J Clin Microbiol,1992,30(3):545-551.
[5]Hall BG.Comparison of the accuracies of several phylogenetic methods using protein and DNA sequences[J].Mol Biol Evol,2005,22(3):792-802.DOI:10.1093/molbev/msi066
[6]Hao M,Zuo L,Shu LP,et al.Sequencing and phylogenetic analysis of E and NS1genes in two strains of dengue type 2[J].Chin J Microbiol Immuno,2004,24(4):287-291.(in Chinese)郝牧,左丽,舒莉萍,等.两株登革2型病毒E、NS1蛋白基因序列测定及其系统发生分析[J].中华微生物学和免疫学杂志,2004,24(4):287-291.
[7]Gubler DJ.Dengue and dengue hemorrhagic fever[J].Clin Microbiol Rev,1998,11(3):480-496.
[8]Wu SG,Hong RT,Ou JM,et al.Characterization of dengue fever cases in Fujian Province,2010[J].Chin J Zoonoses,2011,27(8):763-764.(in Chinese)吴生根,洪荣涛,欧剑鸣,等.福建省2010年登革热特征分析[J].中国人兽共患病学报,2011,27(8):763-764.
