四种de novo组装软件对柞蚕微孢子虫全基因组组装结果的比较
2012-01-18刘宗林许金山周泽扬
刘宗林 许金山* 周泽扬 ,3
(1.重庆师范大学 重庆市动物生物学重点实验室,重庆 400047;2.重庆师范大学 活性物质生物技术教育部工程研究中心,重庆 400047;3.西南大学 家蚕基因组生物学国家重点实验室,重庆 400716)
微孢子虫(Microsporidia)是一类专性寄生于细胞内的单细胞真核生物,可以感染包括昆虫、鱼类和人类在内的几乎所有的动物[1]。感染柞蚕的柞蚕微孢子虫(Nosemaantheraeae),是柞蚕微粒子病的病原,其可经母蛾传染子代,给柞蚕养殖业生产造成严重损失[2]。目前对柞蚕微粒子病的防治主要是通过胚种检疫杜绝母体传染和卵面与养蚕环境消毒。从分子水平探究柞蚕微孢子虫对柞蚕的侵染机制,将有助于建立高效、准确的病害检疫和防治方法。ABYSS、Velvet,SOPAdenovo和Ray是目前常用的四种de novo组装拼接软件。Abyss[3]是基于Bruijn算法的拼接软件,Abyss拼接的优点是可以进行平行运算,并且可以多线程以及多个库同时运行,它往往适用于大基因组的短paired-end reads。有研究者曾利用Abyss对非洲男性的基因组测序得到的35亿对reads进行组装,获得了覆盖度达到了68%的全基因组框架图谱[4]。SOAPdenovo[5]是华大基因开发的高通量从头测序软件,能构建人类基因组大小的从头拼接草图。SOAPdenovo使用的Bruijn算法,是专门为处理Illumina GA产生的短reads而开发的,其为构建参考基因组提供了新的途径[6]。Velvet[7]是一款常用的基因组拼接软件,采用的也是Bruijn算法,它能同时支持fasta、fastq格式的数据,同时支持多个文库的数据同时使用。Velvet仅仅利用短reads和paired-ends信息,就可以产生可观长度的重叠群,Vevlet是目前短序列组装的应用较多的软件,对细菌基因组十分适合。其工作的一般过程简化为:输入短read序列,排除错误,产生高质量的contigs。然后用成对reads信息,检索contigs之间的重复区域[8-9]。Ray[10]是需要使用MPI2.2来进行de novo组装的软件。Ray是构建在RayPlatform基础上的组装软件,可以进行平行运算以及多个库同时运行。它可以同时拼接不同测序软件的reads,在对应的软件下面有重要的参数说明。Ray可以同时拼接Roche/454和Illumina这三种测序工具测序得到的三种不同的基因组序列,结果表明这种混合拼接技术可以大大的减少拼接结果的错误[11]。目前本研究团队完成了柞蚕微孢子虫的染色体核型分析[12],同时也初步完成了柞蚕微孢子虫全基因组的测序,但对数据有效全基因组组装,还处于空白。昆虫微孢子虫基因组一般具有大量的重复序列[13-14],加大了组装的难度,所以组装软件和组装参数的选择显得十分重要。本文将通过比较不同的组装软件和组装参数对柞蚕微孢子虫的从头组装结果,通过比较N50的长度,contig的条数,contigs的总长度等来评价组装结果,从中选择最优的组装软件和组装参数。
1 材料和方法
1.1 基因组数据来源
实验材料来源于重庆师范大学资源昆虫及其病原微生物学研究室测序完成柞蚕微孢子虫基因组数据。
1.2 组装关键参数选择
四种组装软件 Velvet(1.2.07)、Abyss(1.3.4)、SOAPdenovo(v1.04linux 32)Ray(v2.0.0)组装时涉及到的参数很多,但是k-mer是其中最为关键的参数之一,因此,本实验设计了不同的k-mer进行组装结果的比较,而其他参数设为默认。通过perl程序来测试不同的k-mer值.对于Velvet,Abyss,SOAPdenovo,Ray四种软件我们也都通过编写perl脚本程序,完成其他统计分析。
2 结果分析
2.1 不同的k-mer下基因组组装质量比较
采用四种软件组装后,结果显示随着k-mer的增加,组装所得到的基因组全长也是逐渐增加的,最大基因组总长度在5~6M之间(如图1a所示)。我们进一步考察了四种软件在各自不同k-mer值下最大的contig长度,如图1b所示,对于Ray和SOAPdenovo两种软件而言,当k值设定在21~31的范围内,k-mer对最长的contig大小影响不大。而对于Abyss来说,当k=23时最长的contig大小明显变大,而k>23时这种变化趋稳。对于Velvet来说,不同的k值对最长contig大小影响最为显著,具体来说,当k=25时最长contig大小明显高于k-mer=21和k-mer=23,而在k=31时最为显著。而比较四种软件在k-mer数值最优化后组装的最长contig长度,Velvet所获得的结果明显优于另外三种软件。
N50是反映拼接效果最重要的参数,N50越大表明组装结果也越好。如图1c所示,在不同的k-mer下四种软件得到的N50差别很大,N50Velvet>N50Abyss>N50Ray>N50SOAPdenovo。从图中我们可以看出,在设定的k-mer范围内N50Velvet总体上时随着k-mer的增加而增加的,而N50Abyss是先增加后减小,在k-mer=25时最大,而N50Ray、N50SOAPdenovo却没有明显变化。Velvet的N50最大达到了47295bp,明显高于其他三种软件,而N50SOAPdenovo最小,小于1000bp。由此可见不同的k-mer值对不同软件的拼接结果有显著的影响,综合以上分析,我们列出了不同软件在最优参数选择下的组装质量结果,如表1所示。

图1 不同组装软件下基因组组装质量
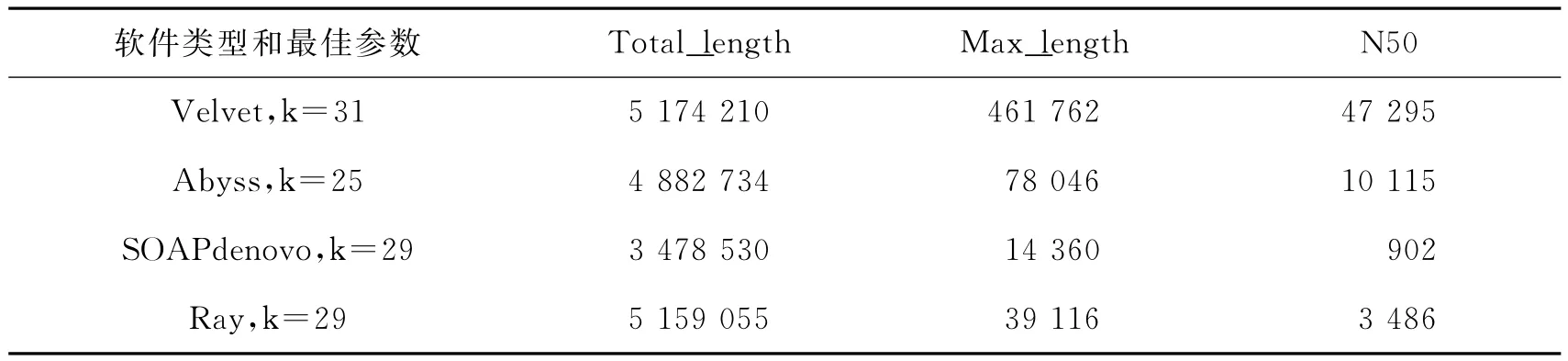
表1 四种软件最优k-mer化的拼接结果比较
2.2 四种软件的硬件资源消耗
我们比较了四种软件在最优组装条件下的系统资源消耗情况,如图2a、图2b所示,Velvet对CPU的消耗是相对小的,而内存的消耗相对要高一点。而Abyss对硬件的消耗相对是最小的。拼接速度上来讲,Abyss、Velvet速率相对较快,而Ray拼接最慢。
通过上面的分析可知,对于柞蚕微孢子虫的基因组,通过比较N50的长度,最大的contigs大小,contigs的总长度等参数得知,Velvet的拼接结果相对最好,最适合柞蚕微孢子虫的基因组组装。在硬件消耗方面Velvet是最高的,但是拼接速度是最快的。

图2 四种软件的硬件资源消耗
3 讨 论
本研究通过比较分析可知,不同的拼接软件对柞蚕微孢子虫的组装结果差别非常大。其中Velvet软件更适合进行柞蚕微孢子虫的基因组组装。究其原因分析可能是由于柞蚕微孢子虫基因组自身的结构特殊性,即包含大量的重复序列,导致不同拼接的结果差异比较大,所以合理的选择拼接软件和参数对于拼接昆虫微孢子虫基因组十分重要的。通过本次研究,对今后柞蚕微孢子虫基因组的分析具有十分重要的理论和实践意义。
[1] Keeling P.Five questions about Microsporidia[J].PLoS Pathog,2009,5(9):e1000489.
[2] 中国农业科学院蚕业研究所.中国养蚕学[M].上海:上海科学技术出版社,1990:847-851.
[3] http://www.bcgsc.ca/platform/bioinfo/software/abyss.
[4] Simpson J T ,Wong K,Jackman S D.Abyss:aparallel assembler for short read sequence data[J].Genome Res 2009,19:1117-1123.
[5] http://soap.genomics.org.cn/soapdenovo.
[6] Li R Q,Zhu H M,Ruan J,Qian W B,et al.Denovo assembly of human genomes with massively parallel short read sequencing[J].Genome Res 2009,20:265-272.
[7] http://www.ebi.ac.uk/~zerbino/velvet/.
[8] Zerbino D R,Birney E.Velvet:algorithms for de novo short read assembly using deBruijn graphs[J].Genome Res 2008,18:821-829.
[9] Zerbino D R.Using the Velvet de novo assembler for short-read sequencing technologies[M].Curr Protoc Bioinformatics 2010.
[10]https://github.com/sebhtml/ray.bastien.
[11]Se′bastien boisvert F J,L.Aviolettel,Corbeil J.Ray:Simultaneous Assembly of Reads from a Mix of High-Throughput Sequencing Technologies[J],Computational Biology,2010,(17):1519-1533.
[12]Xu J S,Wang L J,Zhou Z Y.The Nuclear Apparatus and Chromosomal DNA of the Microsporidian Nosema antheraeae[J].Eukaryotic Microbiology 2011,58(2):178-180.
[13]Xu J S,Wang M,Zhang X Y,et al.Identification of NbME MITE families:Potential molecular markers in the microsporidia Nosema bombycis[J],Invertebrate Pathology,2010,(103):48-52.
[14]Cornman R S,Chen Y P,C.Schatz M C,Street C,Zhao Y,et al.Genomic Analyses of the Microsporidian Nosema ceranae,an Emergent Pathogen of Honey Bees[J],PLoS Pathogens,2009,5(6):e1000466.
