重组慢病毒介导的NMD途径因子UPF1和SMG1可诱导干扰细胞株的构建
2012-01-11柴宝峰常文娟申泉王刚
柴宝峰,常文娟,申泉,王刚
(化学生物学与分子工程教育部重点实验室,山西大学 生物技术研究所,山西 太原 030006)
重组慢病毒介导的NMD途径因子UPF1和SMG1可诱导干扰细胞株的构建
柴宝峰,常文娟,申泉,王刚
(化学生物学与分子工程教育部重点实验室,山西大学 生物技术研究所,山西 太原 030006)
无义介导的m RNA降解途径是一个比较完善的异常mRNA的降解机制,结合在外显子拼接复合体上的多种蛋白决定NMD途径对异常转录物的识别和降解的启动,其中UPF1和SMG1发挥主要功能.UPF1是一个RNA解旋酶和RNA依赖的ATP酶;而SMG1具有磷脂酰肌醇激酶活性,负责UPF1的磷酸化.本研究构建了含有UPF1和SMG-1基因发夹结构的诱导开关基因表达干扰质粒.利用慢病毒介导转化哺乳动物细胞HEK293T细胞得到重组病毒,经鉴定后感染细胞AD_293,目的基因在细胞中得以高效表达.通过继代培养和单克隆化,得到强力霉素诱导干扰UPF1和SMG-1表达的稳定细胞株.
NMD途径;UPF 1;SMG 1;PTC;RNA干扰;shRNA
真核生物细胞拥有多种机制用于保障基因复制的忠实性和表达的准确性.从基因语言到蛋白质语言的转化过程中,经历基因的转录、剪接、修饰、运输和翻译,每一个过程都有一套精密的调控机制.其中无义介导的m RNA降解途径(nonsense-mediated RNA decay,NMD)是一个比较完善的m RNA监控和降解机制,可以选择性地消除含有早发性终止密码子(premature termination codons,PTC)的异常m RNA,结合在外显子拼接复合体(exon-exon junction complex,EJC)上的 UPF1(up-frameshift protein)和SMG1(suppressor with morphological effect on genitalia protein)决定NMD途径对异常转录物的识别和降解途径的启动[1-3].含有PTC的m RNA表达出C端截短的蛋白,有些具有显性-负性(dominant-negative)功能,影响其等位基因所表达的正常蛋白的功能;有些则产生功能获得性(gain-of-function)蛋白,导致机体病变[4].另外,NMD途径还参与调解各种细胞过程中正常的mRNA的表达,诸如细胞分化、胁迫反应和染色体结构和功能的维持以及胚胎发育等[2,5-6].该途径与人类疾病的发生密切相关,尤其是由单基因无义突变导致的各种疾病,如地中海贫血症、血友病、杜氏肌肉营养不良症(Duchenne’s muscular dystrophy,DMD)和囊性纤维病(cystic fibrosis,CF)等[4,7-11].此外,抑癌基因无义突变是导致肿瘤发生的一个重要机制,含有 PTC的抑癌基因(tumor suppressor gene)的转录产物也是 NMD 途径的靶标[8,12-13].因此,对 NMD 途径机制的深入研究具有重要的科学意义和应用前景.本文构建了引发NMD途径的关键因子UPF1和SMG-1的开关型干扰质粒,用慢病毒介导的方法转化哺乳动物细胞,筛选得到了强力霉素诱导干扰UPF1和SMG-1表达的细胞稳定株,为深入探讨NMD途径的机制奠定了基础.
1 材料和方法
1.1 材料
1.1.1 细胞株
大肠杆菌(Escherichiacoli)DH5α为本实验室保存;人胚肾细胞(HEK293T)和AD_293细胞购自Stratagene公司,本实验室保存.
1.1.2 试剂和工具酶
DNA回收试剂盒 (Gel Extraction Mini Kit)购自Bio MIGA公司;质粒抽提试剂盒(Plasmid Mini Kit)购自OMEGA公司;EasyTaqDNA聚合酶购于北京全式金生物技术有限公司;D-MEM细胞培养基和Lipfectinamine2000脂质体转染试剂盒购自Invitrogen公司;胎牛血清购自杭州四季青公司;碱性磷酸酶(Calf intestinal Alkaline Phosphatase)、各种工具酶、T4多核苷酸激酶(polynucleotide kinase)购自TaKaRa公司;T4 DNA ligase购自Promega公司.PCR引物合成由华大基因公司完成;DNA序列测定由北京奥科生物公司完成.
1.2 方法
1.2.1 干扰序列的设计和合成
为了干扰UPF1和SMG-1基因的表达,设计含有发夹结构的寡核苷酸链(sh RNA),在其两侧分别含有一个BglII和KpnI的粘性末端.如表1所示,设计发夹结构分别为对照sh RNAs、与UPF1互补的sh RNA、与SMG-1互补的shRNA.

表1 本研究中所用到的寡核苷酸序列Table 1 Oligonucleotides list used in this study
1.2.2 中间载体的构建
将备用的载体p FRT-U6tet O经BglII和KpnI双酶切,用小牛肠道磷酸酶孵育1 h去磷酸化,琼脂糖凝胶电泳分离、纯化.用T4多核苷酸激酶分别将合成的寡核苷酸链磷酸化.具体操作体系:10μmol/L寡核苷酸链,1 mmol/L三磷酸腺苷,1× 多核苷酸激酶缓冲液,5 U多核苷酸激酶,总体积20μL,37℃孵育1 h.将磷酸化的寡核苷酸链分别混匀,75℃退火10 min,冷却到室温备用.将上述产物(寡核苷酸链)稀释25倍(终浓度为200 nmol/L)与酶切后的质粒进行连接反应.将纯化的质粒片段1μL、退火产物3μL、2×T4 DNA连接酶缓冲液5μL、T4 DNA连接酶1μL混合,总体积10μL.室温孵育1 h.将连接产物分别转化感受态大肠杆菌DH5α.涂板,37℃过夜孵育,挑取阳性克隆,经液体培养后提取的重组质粒p FRT-shC、p FRT-sh U和p FRT-shS(表2,P378).构建的重组质粒经PCR和测序鉴定.
1.2.3 干扰载体p TIG-U6tet Osh的构建
设计引物 BF211/212(表1),分别以构建好的质粒p FRT-sh C、p FRT-sh U、p FRT-shS为模板进行PCR扩增.经NotI和SphI双酶切连接到p TIG慢病毒表达载体上.转化大肠杆菌筛选阳性克隆、提取质粒后经双酶切和测序鉴定.得到重组质粒p TIG-U6tetO-shC、p TIG-U6tet O-sh U和p TIG-U6tetO-shS(表2).
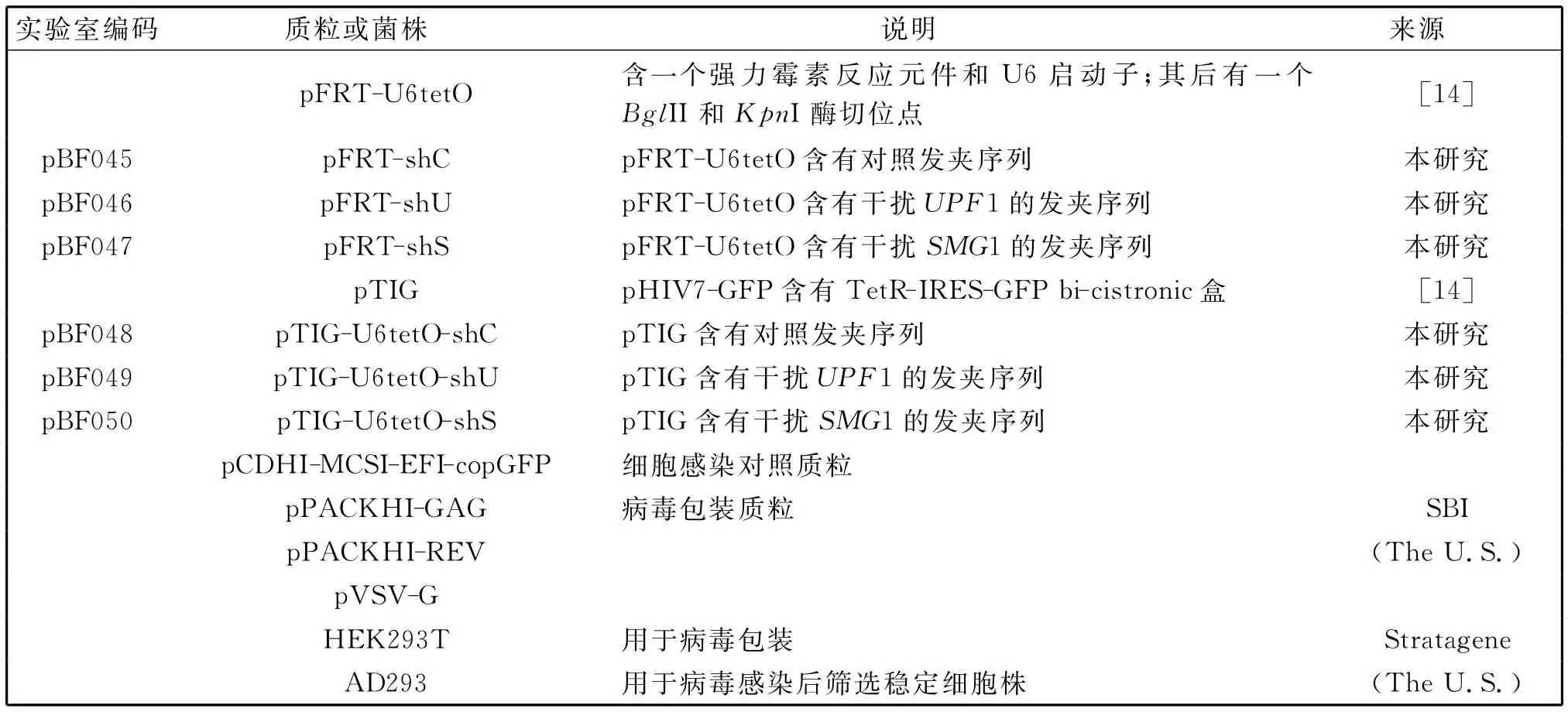
表2 本实验构建和所用的质粒和细胞株Table 2 Plasmids and cell strains used in this study
1.2.4 重组慢病毒的构建
取对数生长期细胞HEK293T,用质量浓度为0.025%的胰酶消化,在底面积为75 cm2的细胞培养瓶中调整细胞浓度为(3.75×106)个,细胞融合度为50%-70%时,参考脂质体转染说明书用阳离子脂质体Lipfectinamine2000分别将重组质粒p TIG-U6tetO-shC、p TIG-U6tetO-sh U 和 p TIG-U6tet O-sh与慢病毒包装质粒pPACKHI-GAG、p PACKHI-REV、p VSV-G共同转染细胞.转染后8 h换液.分别在24 h,48 h和72 h后开始收获细胞培养液上清,3 000 r/min离心5 min沉淀细胞碎片.将上清病毒液用超速离心(50 000×g)在4℃离心90 min浓缩,备用.取80μL病毒悬液,用丙型肝炎病毒核酸检测试剂盒(中山大学,达安基因)反转录病毒总RNA,得到相应的c DNA,用于目的基因的扩增检测.
1.2.5 重组慢病毒感染哺乳动物细胞
取对数生长期AD_293细胞,用质量浓度为0.25%胰酶消化,接种于24孔板,调整细胞浓度为每孔0.6×105-1×105个,将超速离心的病毒沉淀用500μL D-MEM重悬,分别滴入24孔板中.7 d后用激光共聚焦显微镜观察绿色荧光强度,判定细胞的感染效率.筛选稳定细胞株.
2 结果
2.1 慢病毒载体的构建
2.1.1 中间载体的构建
如图1A(P379)所示,在质粒p FRT-U6tet O中有一个强力霉素诱导开关的U6启动子[14].本研究首先将合成的UPF1和SMG-1的发夹结构寡核苷酸进行退火,得到的双链DNA经酶切后连接在U6启动子的下游的BglII和KpnI之间.在质粒的19-36 bp和302-325 bp位置设计引物,对重组质粒进行PCR鉴定.当目的片段连接成功时,引物扩增片段的大小应该是~310 bp,而没有连接成功时应该是~380 bp.结果如图1B所示,泳道1是没有连接目的片段的扩增产物,泳道2、3、4分别为连接了对照序列片段、SMG-1发夹序列片段和UPF1发夹序列片段.得到的中间重组质粒分别命名为p FshC、p FshS和p Fsh U.
2.1.2 慢病毒载体的构建
在质粒p FRT-U6tetO上的19-36 bp和302-325 bp位置设计引物(表1),将含有U6启动子和发夹结构的基因片段扩增出来,连接到p MD-18T载体进行测序.经NotI和SphI酶切后连接到p TIG质粒中,经酶切鉴定(如图2A,P380)后进一步测序证实.得到一个含有U6启动子、干扰发夹序列、Tet R调控序列和GFP基因的基因表达盒(图2B),该表达盒位于慢病毒表达质粒中.质粒分别命名为p TIG-shC、p TIG-sh U、p TIG-shS,分别表达干扰对照序列、干扰UPF1和SMG1的基因片段,该干扰过程受强力霉素的诱导调控.

图1 重组慢病毒中间载体的构建A p FRT-U6tetO质粒载体图谱;B中间载体p FRT-U6tetO的PCR鉴定.Mr:Takara DL500 DNA Marker,泳道1:质粒p FRT空载体的PCR扩增产物;泳道2:重组质粒p FshC的PCR扩增产物;泳道3:重组质粒p FshS的PCR扩增产物;泳道4:重组质粒p Fsh U的PCR扩增产物Fig.1 Construction of mediated recombinant lentivirus vectorsA:the map of plasmid p FRT-U6tetO;B:the PCR analysis of mediated plasmids p FRT-U6tetO.Mr:Takara DL500 DNA Marker;Lane 1:PCR product of plasmid p FRT;Lane 2:PCR product of plasmid p FshC;Lane 3:PCR product of plasmid p FshS;Lane 4:PCR product of plasmid p Fsh U
2.2 干扰细胞株的构建
2.2.1 重组慢病毒制备
将构建的三种重组慢病毒基因表达质粒p TIG-U6tet O-shC、p TIG-U6tet O-shS和p TIG-U6tet O-sh U分别与病毒包装质粒混合转染人胚肾细胞HEK293T(图3A,P380).经培养后,离心收集病毒颗粒.取80μL病毒,经反转录得到病毒的cDNA,用引物BF175/176进行扩增,得到图2所示的基因盒中U6Tet O与Tet R之间的基因片段,总长度为2 438 bp(图3B),并经测序证实.从病毒中克隆到目的基因,说明目的基因片段被成功包装到病毒中,可用于下一步的细胞的感染,构建稳定的细胞株.病毒颗粒分别命名为V-shC、V-shS和V-sh U.
2.2.2 细胞的感染和筛查
培养AD_293细胞,调整细胞浓度为每孔0.6×105-1×105个时,用含有基因表达盒的重组慢病毒V-shC、V-shS和V-sh U分别感染细胞AD_293.培养7 d后在荧光显微镜下观察.如图4(P381)所示,表达基因盒中的eGFP基因作为标记,绿色荧光蛋白的表达说明外源基因在细胞中得到了成功表达,这说明人工构建的慢病毒成功感染了AD_293细胞,得到了含有对UPF1和SMG-1进行可诱导干扰的细胞株.
3 讨论
本研究成功构建了强力霉素诱导表达的用于干扰NMD途径关键因子UPF1和SMG1的重组慢病毒质粒,并制备了相应的重组病毒,感染目的细胞AD_293,绿色荧光蛋白基因在细胞中得以高效表达,说明目的基因已经通过慢病毒成功转染到细胞中.本实验得到的细胞株为下一步研究NMD途径的机制、无义突变基因的表达调控机制奠定了基础.另外,该细胞株可以用于筛选受到NMD途径调控的参与细胞正常生理活动的基因和一些与肿瘤发生相关的基因[12].

图2 重组慢病毒干扰载体的构建A重组慢病毒干扰质粒p TIG-U6tetO-sh的酶切鉴定.重组质粒经的Not I和Sph I酶切电泳.Mr:Trans 2K Plus DNA Marker,泳道1:重组质粒p TIG-U6tetO-shC;泳道2:重组质粒p TIG-U6tetO-shS;泳道3:重组质粒p TIG-U6tetO-sh U;泳道4:质粒p TIG.B强力霉素诱导发夹序列干扰基因表达的基因盒组成结构Fig.2 Construction of recombinant lentivirus interference plasmidA:Enzyme analysis of recombinant lentivirus interference plasmid by Not I and Sph I.Mr:Trans 2K Plus DNA Marker;Lane 1:recombinant lentivirus interference plasmid p TIG-U6tetO-shC;Lane 2:recombinant lentivirus interference plasmid p TIG-U6tetO-shS;Lane 3:recombinant lentivirus interference plasmid p TIG-U6tetO-sh U;Lane 4:control plasmid p TIG.B:map of gene cassette encoding dox-inducible interference hairpin structure
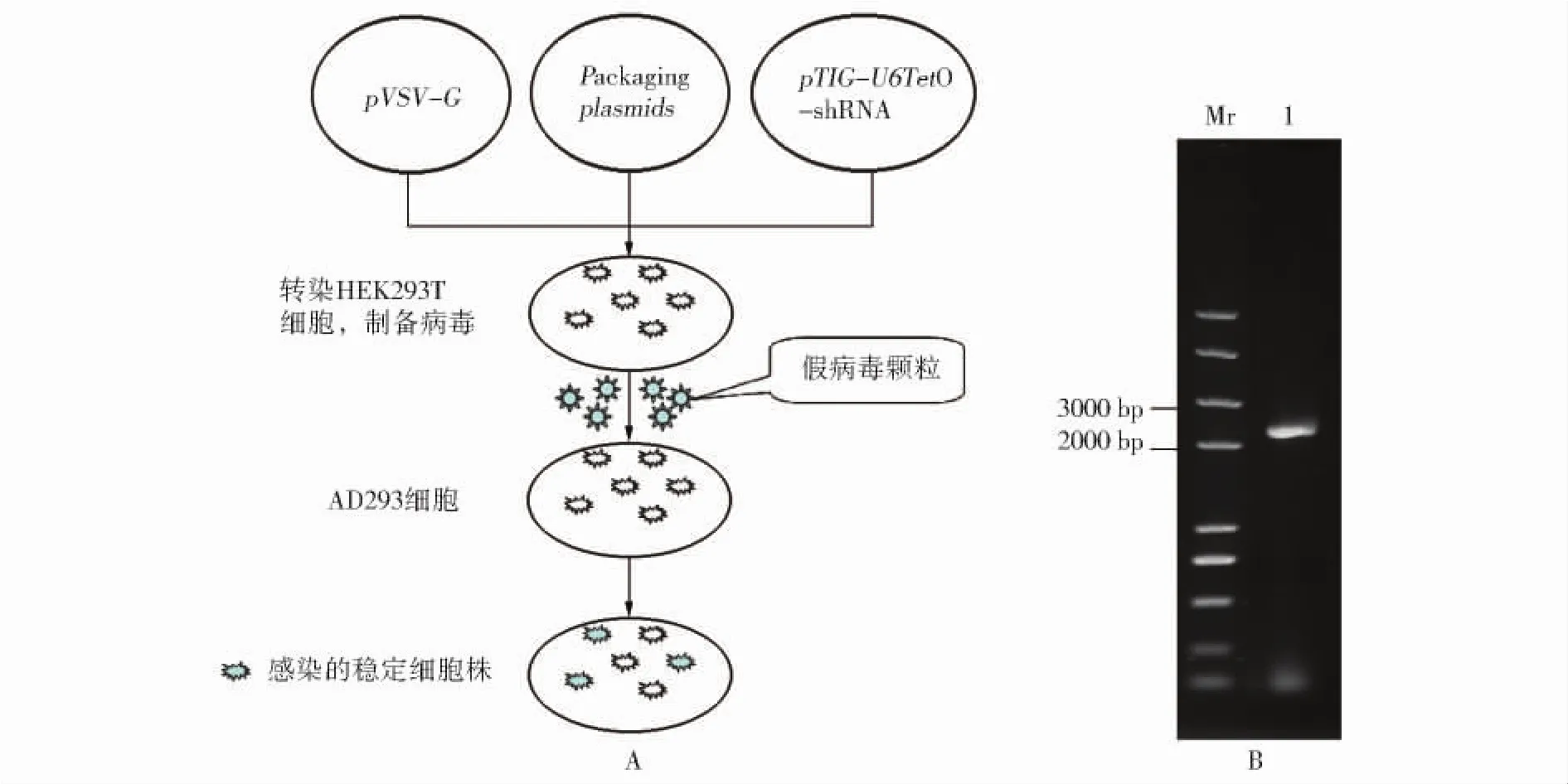
图3 重组慢病毒的制备和鉴定A病毒包装和稳定细胞株制备流程;B从病毒基因组中扩增的目的基因片段.Mr:Trans 2K Plus DNA Marker,泳道1:从病毒cDNA扩增的目的基因片段,长度为2 438 bpFig.3 Preparation and analysis of recombinant lentivirusA:flow-sheet of Preparation and analysis of recombinant lentivirus;B:PCR product from prepared recombinant lentivirus.Mr:Trans 2K Plus DNA Marker;Lane 1:gene fragment amplified from recombinant lentivirus and the full-length is 2 438 bp

图4 重组慢病毒感染AD_293细胞,绿色荧光蛋白在细胞中的表达.图Control:细胞内发夹结构中含有的基因序列与基因组片段没有互补关系;图UPF1:细胞内的基因盒中含有干扰UPF1的发夹结构;图SMG-1细胞内的基因盒中含有干扰SMG1的发夹结构;Fig.4 Expression of green fluorescence in AD_293 tranfected by recombinant lentivirus Fig.Control:gene in hairpin cassette in AD_293 cells have no complementary to genomic DNA of human cell;Fig.UPF1:gene in hairpin cassette in AD_293 cells interfere the expression of UPF 1;Fig.SMG-1 gene in hairpin cassette in AD_293 cells interfere the expression of SMG1
[1]Frischmeyer P A,Dietz H C.Nonsense-mediated mRNA Decay in Health and Disease[J].HumMolGenet,1999,8(10):1893-1900.
[2]Nicholson P,Yepiskoposyan H,Metze S,etal.Nonsense-mediated mRNA Decay in Human Cells:Mechanistic Insights,Functions Beyond Quality Control and the Double-life of NMD Factors[J].CellMolLifeSci,2010,67(5):677-700.
[3]Brogna S,Wen J.Nonsense-mediated m RNA Decay(NMD)Mechanisms[J].NatStructMolBiol,2009,16(2):107-113.
[4]Bhuvanagiri M,Schlitter A M,Hentze M W,etal.NMD:RNA Biology Meets Human Genetic Medicine[J].BiochemJ,2010,430(3):365-377.
[5]Hwang J,Maquat L E.Nonsense-mediated mRNA Decay(NMD)in Animal Embryogenesis:to Die or not to Die,That is the Question[J].CurrOpinGenetDev,2011,21:1-9.
[6]Sharifi N A,Dietz H C.Physiologic Substrates and Functions for Mammalian NMD.In:Nonsense-Mediated mRNA Decay[M].Edited by Maquat LE.Georgetown,Texas:Landes Bioscience,2006:97-109.
[7]Abbas S,Erpelinck-Verschueren C A,Goudswaard C S,etal.Mutant Wilms’tumor 1 (WT1)mRNA with Premature Termination Codons in Acute Myeloid Leukemia(AML)is Sensitive to Nonsense-mediated RNA Decay(NMD)[J].Leukemia,2010,24(3):660-663.
[8]Karam R,Carvalho J,Bruno I,etal.The NMD mRNA Surveillance Pathway Downregulates Aberrant E-cadherin Transcripts in Gastric Cancer Cells and in CDH1 Mutation Carriers[J].Oncogene,2008,27(30):4255-4260.
[9]Holbrook J A,Neu-Yilik G,Hentze M W,etal.NMD and Human Disease[M]//Nonsense-Mediated mRNA Decay.Edited by Maquat LE.Georgetown,Texas:Landes Bioscience,2006:111-119.
[10]James P D,Raut S,Rivard G E,etal.Aminoglycoside Suppression of Nonsense Mutations in Severe Hemophilia[J].Blood,2005,106(9):3043-3048.
[11]Hamed S A.Drug Evaluation:PTC-124-a Potential Treatment of Cystic Fibrosis and Duchenne Muscular Dystrophy[J].IDrugs,2006,9(11):783-789.
[12]Gardner L B.Nonsense-mediated RNA Decay Regulation by Cellular Stress:Implications for Tumorigenesis[J].Mol CancerRes,2010,8(3):295-308.
[13]Wolf M,Edgren H,Muggerud A,etal.NMD Microarray Analysis for Rapid Genome-wide Screen of Mutated Genes in Cancer[J].CellOncol,2005,27(3):169-173.
[14]Aagaard L,Aamarzguioui M,Sun G,etal.A Facile Lentiviral Vector System for Expression of Doxycycline-inducible Sh RNAs:Knockdown of the pre-miRNA Processing Enzyme Drosha[J].MolTher,2007,15(5):938-945.
Construction of Cell Strain Containing Inducible Interference of NMD Factor UPF1 and SMG1 Mediated by Lentiviral System
CHAI Bao-feng,CHANG Wen-juan,SHEN Quan,WANG Gang
(KeyLaboratoryofChemicalBiologyandMolecularEngineering,MinistryofEducation,InstituteofBiotechnology,ShanxiUniversity,Taiyuan030006,China)
Nonsense-mediated decay(NMD)is well known by the lucid definition of being a RNA surveillance mechanism that ensures the speedy degradation of mRNAs containing premature translation termination codons.The definition of aberrant transcription and start of NMD are determined by the protein complex binding on EJC,where UPF1 and SMG1 play key role for NMD path.UPF1 is a RNA helicase and a RNA-dependent ATPase.SMG1 is responsible for UPF1 phosphorylation as a phosphatidylinositol 3-kinase.In this study,we construct the p HIV-7-derived lentiviral vector p TIG (p HIV7-Tet R-IRES-GFP)encoding a U6tet O-promoted short hairpin RNA (sh RNA)cassette containing UPF1 and SMG1.The resultant plasmids were transfected into HEK293T cell with the help of packaging plasmids to result in recombinant virus harboring hairpin RNA cassette.The resultant virus were then used to infect AD_293 cell to observe the expression of hairpin RNA gene.We have obtained the cell strains expressing hairpin RNA of UPF1 and SMG1,and these cell strains could be used for next research on the mechanism of NMD and gene screen.
NMD path;UPF 1;SMG 1;premature termination codon;RNA interference;sh RNA
Q813
A
0253-2395(2012)02-0376-07*
2012-02-16
国家自然科学基金( 31172078;30770294);山西省自然科学基金(2009011040-1);山西省留学归国基金项目
柴宝峰(1967-),男,山西闻喜人,博士,教授,研究领域:分子细胞生物学.E-mail:bfchai@sxu.edu.cn
