微波辅助提取-液相色谱-氢化物发生-原子荧光光谱法分析沉积物中砷的形态
2012-01-11武铄虞锐鹏宋启军
武铄 虞锐鹏 宋启军
(1江南大学化学与材料工程学院;2江南大学国家食品科学与技术重点实验室,江苏无锡 214122)
微波辅助提取-液相色谱-氢化物发生-原子荧光光谱法分析沉积物中砷的形态
武铄1,2虞锐鹏2宋启军1*
(1江南大学化学与材料工程学院;2江南大学国家食品科学与技术重点实验室,江苏无锡 214122)
采用微波辅助提取-液相色谱-氢化物发生-原子荧光光谱法(LC-HG-AFS)联用技术分析了太湖沉积物中砷的形态[亚砷酸(As(III))、二甲基砷酸钠(DMA)、一甲基砷酸二钠(MMA)和砷酸As(V)]。测得沉积物中以无机砷为主,且以As(V)居多。选定以1mol/L的磷酸和0.1mol/L抗坏血酸为提取液,在微波辅助萃取(功率为60W,时间12min)下,萃取率达79.84%~91.57%,回收率在94.78%~107.6%之间。4种砷的形态在0~160μg/L之间时线性良好,检测限为0.6~2.3μg/L,相对标准偏差RSD为1.62%~2.20%。方法具有简便、快速、灵敏的特点。
砷形态;沉积物;液相色谱;原子荧光光谱
1 前言
砷在环境中的移动性、毒性及生物有效性主要取决于其存在形态,因此砷的价态分析对环境效应评价具有重要的意义。土壤中的砷以无机砷为主,有些淤泥和沉积物中含有少量的有机砷,如一甲基砷(MMA)和二甲基砷(DMA),提取方法对于砷的测定至关重要。按照辅助仪器的不同,有水浴[1]、超声[2],震荡提取[3],微波[4]和加速溶剂萃取等[5]。其中微波提取用时短、提取率高、试剂消耗量少、且不破坏元素的原有形态。按照提取剂的不同,有水、甲醇[6]、不同的盐[7]、磷酸[8]提取等。在众多的提取剂中,磷酸的效果最佳。基于此,通过微波辅助磷酸提取法,采用液相色谱-氢化物发生-原子荧光光谱法检测,建立了沉积物中4种砷形态的分析方法,并对太湖沉积物的砷存在情况进行了测定。
2 实验部分
2.1 仪器及试剂
色谱-原子荧光光谱联用仪(AF-610D2,北分瑞利分析仪器公司),工作参数如下:PMT电压320V,主电流/辅电流50/30mA,载气/辅助气流量600/600mL/min,KBH4(3.0%),HCl(10%),HCl流速4mL/min,进样量200μL;
Hamilton PRP-X100阴离子色谱柱(250mm× 4.1mm,10μm)为分离柱(瑞士Hamilton公司),Hamilton PRP X-100阴离子交换色谱柱或等效柱(10mm×4.1mm,10μm)为保护柱(瑞士Hamilton公司);
微波加速反应仪(MARS-5,美国CEM公司);冷冻干燥仪(LABCONCO-Freezone stoppering Tray dryer,USA);电热恒温水浴锅(HWS12,上海一恒科技有限公司)。
亚砷酸钠As(Ⅲ)和砷酸氢二钠As(V)(国家计量科学院),二甲基砷酸钠(DMA)(美国accustandard公司),一甲基砷酸二钠(MMA)(sigma公司)。GBW07409标准土壤(黑龙江省环境保护科学研究院);流动相20mmol/L磷酸盐缓冲溶液用0.45μm水膜过滤;分析纯KBH4溶于KOH(0.2%)作为还原剂;硫脲(10g/L)作为总砷的还原剂。草酸铵、碳酸氢钠和碳酸钠为分析纯,磷酸为优级纯,上述试剂均购于国家药品试剂有限公司。
2.2 样品采集
采用抓斗式底泥采样器采样,清除大颗粒石块和根茎等杂物,置于聚四氟乙烯瓶中保存。回实验室后,于-40℃冰箱中冷冻24h,取一定量的沉积物置于冷冻干燥仪中冷冻干燥48h。冻干后的样品用粉碎机粉碎,过0.15mm筛,低温干燥后备用。
2.3 样品总砷的提取
称取大约0.3g样品于聚四氟乙烯罐中,加5mL浓硝酸浸泡过夜,次日加入5mL水,微波消解。消解好的样品置于电炉上赶酸至0.5mL后定容于25mL的容量瓶中。取10mL该消解液置于25mL容量瓶中,加入5mL盐酸(1+1)和5mL硫脲,用超纯水定容后分析。
2.4 样品砷形态的提取
微波提取:于消解罐中加入0.3g样品以及10mL磷酸提取液(1mol/L)以及抗坏血酸(0.1mol/L),进行微波辅助提取。然后将消解液转入离心管中,残渣再次加入磷酸提取。合并两次消解液,在4000r/min下离心15min,上清液定容于50mL容量瓶中,待分析。
3 结果与讨论
为了保证提取前后形态不发生变化,实验选择了温和的提取条件。这是由于磷和砷处于同一主族,有着相似的化学性质。土壤中砷主要是被Fe、Mn的氢氧化物胶体吸附,这样通过离子交换的原理,磷同砷就形成竞争吸附,从而提取出吸附在固体中不容易溶出的砷,达到较高的提取率[9],所以选取磷酸作为提取液。
3.1 提取液的优化
为了防止提取过程中As(III)被氧化成As(V),根据文献在提取液中加入了0.1mol/L抗坏血酸[10]。通过加标回收得知,直接用磷酸提取时,90%As(III)转化为As(V),而加抗坏血酸后,回收率在95.4%~107.9%之间,说明用该法是可行的。浓度是影响提取率的重要因素。当磷酸浓度从0.3mol/L提高到1mol/L时萃取率明显升高。但是当浓度升高到3mol/L时,标准土壤As(V)的峰形严重展宽,且As(III)与DMA部分重合,故选择提取液浓度为1mol/L。
3.2 微波提取实验的条件优化
研究了微波功率与时间对提取率的影响,选取了30~75W之间的功率,在30W时功率太低,基本不能满足要求,在75W时,温度升高,磷酸在高温高压下会有爆炸的危险。故选择了45和60W实验分析,结果见表1。可以看出在45W时,增加时间,最大的提取率仅为68%,且达到30min时,样品出现焦灼。在60W,随着提取时间的延长,提取率明显提高,在18min时,提取率约为90%,但此时As(V)的峰有展宽现象。再考虑到用时问题,选择了60W,12min作为最终的提取条件。

表1微波提取条件实验Table 1 The experiment conditions for microwave-assisted extraction μg/g
3.3 As(III)在土壤及沉积物中的稳定性实验
由于As(III)容易被氧化,所以样品的稳定性对形态分析至关重要。Pizarro[1]等对土壤提取液的稳定性进行了考察,发现在冰箱(4℃)保存的条件下,30d内形态不发生变化,90d后As(III)才全部氧化成As(V)。而Gallardo[11]报道,As(III)在保存20min即开始发生转化。这些差异可能是由于土壤基质和pH值不同所导致,但都表明了As(III)形态易发生变化。考察了浓度和pH值对氧化速率的影响。发现将提取好的试液稀释10倍,As(III)6h内不发生转化。用氨水调节试液pH值为7,As(III)的浓度在6h内可保持稳定。这个过程中虽然会有白色沉淀产生,但过滤后不影响测定。由此可见,As(III)的氧化速率可以通过浓度和pH值来控制。将提取好的溶液,置于冰箱中保存(4℃),并在不同时间进行测定,结果见图1。可见试样在6h内基本稳定,而在60h后As(III)已全部氧转化为As(V),故在6h内完成测试对形态影响不大。
3.4 校准曲线、检出限以及精密度
浓度为50μg/L的混合标样测试图(图2)。可以看出加入提取液后As(V)峰型展宽,但并不影响其准确定量。以浓度为横坐标,峰面积为纵坐标,绘制线性曲线,并进行回归方程计算,结果见表2。重复5次浓度为50μg/L的标准溶液,计算RSD。检出限的计算:DL=3S/N(其中N基线噪声;S仪器的灵敏度元)。

图1 土壤提取后砷形态的稳定性实验Figure 1.Stability test for arsenic species after being extracted from soil.

表2 线性方程、检出限、相对标准偏差Table 2 Linear equation,limit of detection,relative standard deviation
3.5 实际样品的测定
对环太湖的十五个点位(见图3a)采集到的沉积物进行了测定。图3b作为一个例子给出了样品及其加标图。总砷的浓度范围在8.369~13.07μg/g,提取率的范围在79.84%~91.57%之间。沉积物中无机砷是主要存在的形态,且以As(V)为主,两种有机砷MMA、DMA均未检出。为了验证方法的可靠性,并且观察提取过程中形态是否发生变化,采用加标回收对前处理方法进行评价。选取测得砷含量较高(椒山土壤)和较低(平台山土壤)的两个地点进行加标回收。由表3可知,回收率As(III)为94.90%~107.6%,As(V)为96.00%~103.35%,MMA为94.26%~98.90%,DMA为94.78%~103.4%。为了进一步验证方法的精密度,表4列出了混合标准溶液多次测定的RSD,均小于3%。Ellwood等[12]分析了澳大利亚Macquarie湖23个点位的沉积物,发现无机砷也是主要存在的形态,As(V)的含量较高,浓度范围为0.11~5μg/g,As(III)为0.23~2.43μg/g,总砷的浓度基本都在7μg/g左右,仅有两个点位较高,分别为20.20、15.74μg/g。Hasegawa[13]等报道了日本Kiba湖砷的含量As(III)未检出~4.1μg/g,As(V)2.3~7.6μg/g,DMA未检出~2.5μg/g。可以看出As(V)是湖泊中主要的污染形态,与上述报道相比,太湖中砷各个形态的浓度略高。

图2 优化条件下浓度为50μg/L四种砷化合物混合标准溶液色谱图Figure 2.Chromatogram of the mixed arsenic standards(50μg/L).

表3 样品的加标回收实验Table 3 Recovery studies of spiked sediment samples(n=3) ng/g
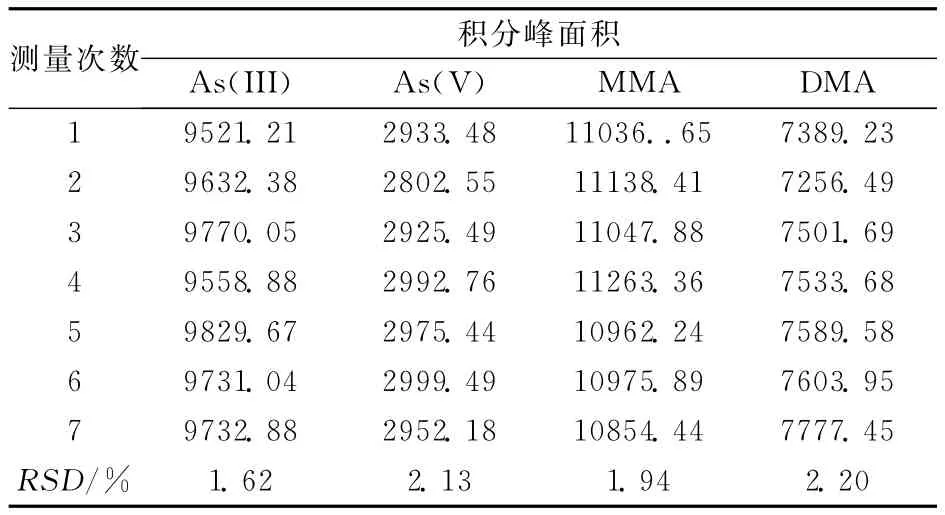
表4 40μg/g的混合标准溶液测试结果(n=7)Table 4 The testing results for the mixed standard solution with concentration of 40μg/g by seven times injection

图3a 太湖水样采集点分布图Figure 3a.The selected sampling positions in the Tai lake.
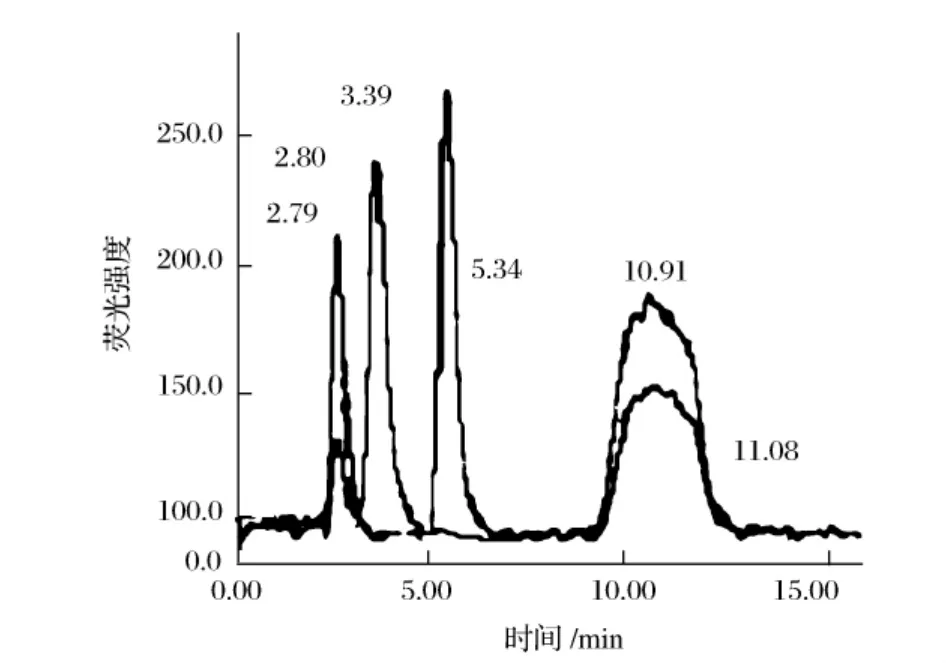
图3b 平台山样品及其加标图Figure 3b.XFS spectra for the sediment sample collected from Pingtai mountain before and after standard addition.
4 结论
采用了液相色谱-氢化物发生-原子荧光光谱法分析太湖沉积物中砷的污染情况。以磷酸和抗坏血酸为提取液,在微波辅助的条件下提取,回收率较高。方法简单方便快速,提取率高,大大缩短了提取时间,且形态提取后无变化,是值得推广的一种方法。
[1]张静,刘晓端,江林.土壤中不同形态砷的分析方法[J].岩矿测试,2008,27(3):179-183.
[2]Bohari Y,Lobos G,Pinochet H et al.,Speciation of arsenic in plants by HPLC-HG-AFS:extraction optimisation on CRMmaterials and application to cultivated samples[J].J.Environ.Monit,2002,4:596-602.
[3]Pongratz R.Arsenic speciation in environmental samples of contaminated soil[J].The Science of the Total Environment,1998,224:133-141.
[4]Garcia-Manyes S,Jiménez G,PadróA,et al.Arsenic speciation in contaminated soils[J].Talanta,2002,58(1):97-109.
[5]Carabias-Martínez R,Rodr′ıguez-Gonzalo E,Revilla-Ruiz P,et al.Pressurized liquid extraction in the analysis of food and biological samples[J].Journal of Chromatography A,2005,1089(1-2):1-17.
[6]Pizarro I,Gómez M,Cámara C,et al.Arsenic speciation in environmental and biological samples Extraction and stability studies[J].Analytica Chimica Acta,2003,495:85-98.
[7]Vassileva1E,Becker A,Broekaert J A C,Determination of arsenic and selenium species in groundwater and soil extracts by ion chromatography coupled to inductively coupled plasma mass spectrometry[J].Analytica Chimica Acta,2001,441:135-146.
[8]Orero Iserte L,Roig-Navarro A F,Hern′andez F,Simultaneous determination of arsenic and selenium species in phosphoric acid extracts of sediment samples by HPLCICP-MS[J].Analytica Chimica Acta,2004,527:97-104.
[9]Bissen M,Frimmel F H.Fresenius,Speciation of As(III),As(V),MMA and DMA in contaminated soil extracts by HPLC-ICP/MS[J].Anal Chem,2000,367:51-55.
[10]Georgiadis M,Cai Y,Solo-Gabriele H M.Speciation analysis of arsenic in environmental solids Reference Materials by high-performance liquid chromatographyhydride generation-atomic fluorescence spectrometry following orthophosphoric acid extraction[J].Environmental Pollution,2006,141:22-29.
[11]Vergara Gallardo M,Bohari Y,Astruc A,et al.Speciation analysis of arsenic in environmental solids Reference Materials by high-performance liquid chromatography-hydride generation-atomic fluorescence spectrometry following orthophosphoric acid extraction[J].Analytica Chimica Acta,2001,441:257-268.
[12]Ellwood MJ,Maher WA.Measurement of arsenic species in marine sediments by high-performance liquid chromatography-inductively coupled plasma mass spectrometry[J].Analytica Chimica Acta,2003,477:279-291.
[13]Hasegawa H,Rahman MA,Matsuda T,et al.Effect of eutrophication on the distribution of arsenic species in eutrophic and mesotrophic lakes[J].Science of the Total Environment,2009,407:1418-1435.
Speciation of Arsenic in the Sediments of Tai Lake by Microwave-Assisted Extraction High Performance Liquid Chromatography and Hydride Generation-Atomic Fluorescence Spectrometry
WU Shuo1,2,YU Ruipeng2,SONG Qijun1*
(1.SchoolofChemicalandMaterialEngineering,JiangnanUniversity;2.StateKeyLaboratoryofFoodandScienceandTechnology,JiangnanUniversity,WuXi,JiangSu214122,China)
Based on microwave-assisted extraction-liquid chromatography and hydride generation-atomic fluorescence spectrometry,an analytical method for speciation of arsenic containing compound(arsenite As(III),dimethylarsinic acid(DMA),monomethylarsonic acid(MMA)and arsenate As(V))in the Tai lake sediment was developed.The results showed that the main speciation in sediments is inorganic arsenic and arsenate As(V)is the majority of the speciation.This paper chose 1mol/Lphosphoric acid as extracting solution and the microwave-assisted extraction was conducted for 12mins with a power of 60W.The extraction rates were in the range of 79.84%~91.57%.The linear responses for four arsenic species were in the range of 0~160μg/Lwith the limits of detection in the range of 0.6~2.3μg/L,and the recovery rates in the range of 94.78%~107.6%.The developed method is simple,rapid and sensitive,and may be applicable for arsenic speciation in other sediments or soil samples.
arsenic speciation;sediment;LC;AFS
*通讯作者:宋启军,教授,博士生导师。E-mail:qsong@jiangnan.edu.cn。
O657.31;TH744.16
A
2095-1035(2012)01-0022-05
10.3969/j.issn.2095-1035.2012.01.0004
2011-09-22
2011-12-30
国家自然科学基金项目(20977042)。
武铄,女,硕士研究生,环境分析化学方向。E-mail:wushuo1987@163.com。
