兔子朊蛋白结构稳定性的分子动力学研究
2012-01-08金相逢刘亚芳张敬来
陈 欣,金相逢,刘亚芳,张敬来
(河南大学 化学化工学院,河南 开封475004)
兔子朊蛋白结构稳定性的分子动力学研究
陈 欣,金相逢,刘亚芳,张敬来*
(河南大学 化学化工学院,河南 开封475004)
朊蛋白病是一种能够对人类和动物带来致命影响,并具有高度传染性的神经退行性疾病.兔子是目前已经报道的哺乳类动物中对朊蛋白病免疫的少数几个物种之一.我们将分子动力学和操控式分子动力学模拟相结合,研究了兔子正常朊蛋白的结构稳定性;同时讨论了蛋白结构的收敛性及刚性分布,并揭示了兔子朊蛋白中关键二级结构的动力学以及受力各向异性特征,证实了兔子朊蛋白结构的稳定性特征.
兔子朊蛋白;结构稳定性;分子动力学模拟;操控式分子动力学模拟
Prion diseases,known as transmissible spongiform encephalopathies(TSEs)associated with Creutzfeldt-Jackob disease(CJD),Kuru in humans,scrapie in sheep and bovine spongiform encephalopathy(BSE),are invariably fatal and highly infectious neurodegenerative diseases[1-3].Acquiring structural information of normal cellular prion protein(PrPc)in scrapie isoform (PrPsc)is essential for understanding the pathogenesis of prion disease.The amino sequences of PrPc and PrPsc are the same,but the secondary structure of the two isoforms is quite different:PrPc is rich inα-helical structure,whereas PrPsc contains a significant increase inβ-sheets content(from 3%to 45%)[4].Thus the study of the structural dynamics mechanism is of great importance for understanding the pathogenesis of prion diseases.
Recently,both experimental and computational studies have been applied to reveal the mechanism of structural transformation from PrPc to PrPsc.X-ray diffraction (XRD),nuclear magnetic resonance(NMR)and electron microscopy(EM)analyses have been used to elucidate the structure of protein[4-6].However,it is difficult to probe the dynamics and flexibility of protein.Molecular dynamics(MD)simulation has been verified to be one of the most efficient computational methods to provide continuous dynamic information on proteins at atomic level[7].Nevertheless,the folding and unfolding time of protein usually varies from 10μs to 1ms[8],which means it is difficult to observe the folding and unfolding processes of protein by pure MD simulation.For example,recent simulations have shown that the structure of prion protein is convergent and it is hard to observe the unfolding pathway[9].Fortunately,steered molecular dynamics(SMD)simulation as an effective and powerful method to probe the mechanical properties of biomolecules can be adopted to quickly probe the dynamic characteristics of protein;and SMD method has become a powerful tool for complementing in vitro single-molecule experiments[10].
Bearing those perspectives in mind and noticing that rabbit is one of the several mammalian species reported to be resistant to infection from prion diseases,in the present research we combine MD simulation with SMD simulation to study the thermo-stability and the interaction mechanism of dynamic process of normal rabbit prion protein(rPrPc).Thus rPrPc was selected as an example to study the structural stability of prion protein.The flexibility of the key secondary structure and the distinct interaction of the protein,as well as the force response of rPrPc were investigated.This research,hopefully,is to help to promote the study of pathogenesis and drug design of prion diseases.
1 Methods
All MD and SMD simulations were performed with NAMD version 2.7[11]of Charmm 27force field[12].The NMR structure of rPrPc was cited from Protein Data Bank (PDB)with PDB ID code 2FJ3[13].The protein was purified from the initial structure and the missing atoms were added.Then protein was immersed in a periodic water box with TIP3Pmodel water molecules.The water box was designed to be large enough to accommodate the structure changes during MD and SMD processes.The volume of the water box is 4.739×6.096×4.352nm3and the number of water molecules is 3 238.The long-range electrostatic interactions were calculated using the Particle Mesh Ewald(PME)summation scheme.All simulations were carried out with a time step of 2fs.Both the van der Waals and columbic interactions were truncated at 1nm.NPT ensemble was used and Langevin method was employed to control the constant temperature at 310Kand constant pressure at 101.3kPa during the MD process.Energy minimization was performed to optimize the geometry of rPrPc and then MD simulation was applied to equilibrate the system for 10ns.
In association with the MD simulation,a series of SMD simulations were carried out to investigate the dynamics of rPrPc.In principle,SMD simulation is applied by attaching a harmonic restraint to one or more atoms in the system,and then either the stiffness or the position of the restraint is changed to pull the atom along.Constant velocity pulling(PCV)method is one of the basic operations of SMD,and it was used in this research to disturb the rPrPc system with external forces.In the PCV simulation,the SMD atoms are attached to a dummy atomviaa virtual spring and this dummy atom moves at a constant velocity in desired direction.Since the protein could be roughly described in a rectangular box,the six faces of the box were selected as the six different starting orientations of SMD directions.The initial orientation of rPrPc was denoted as+x,and the other five orientations were denoted as-x,+y,-y,+z,and-z,respectively.In SMD simulations,a slab of water molecules with a thickness of 0.3nm was tagged as the SMD atoms and the external force was applied to the SMD atoms along the selected directions;and the other water molecules and rPrPc were free.As shown in Fig.1,SMD waters are tagged in-xdirection and would be pulled along+xaxis to disturb the protein.In SMD simulations,all the parameters were carefully adjusted according to the rigidity of protein itself.The spring constantkwas set to be 12 558kJ·mol-1·nm-2and the pulling velocity was fixed at 5×10-5nm·fs-1to obtain good SMD observation window.The pulling distance was set to be 4-5.7nm according to the system size and the periodic boundary conditions.One thousand frames of trajectories were deposited during the SMD simulation.
2 Results and discussion
2.1 MD simulation results

Fig.1 The snapshot of the rPrPc system with SMD waters tagged in-xdirection.SMD water molecules are marked in red and white with VDW model.The free water molecules are shown with the line model,and rPrPc was displayed with the cartoon model
MD simulation is essential to achieve equilibrium state,and two criterions,i.e.,the potential energy of protein and the root mean square deviation (RMSD),are generally adopted to judge whether it has achieved equilibrium or not during the simulation procedure[14].Since the energy expression of rPrPc immersed in water box is complicated,the potential energy and RMSD of PrPc were extracted individually from the simulation system so as to describe the final state of rPrPc more exactly.
The potential energy of rPrPc during the 10ns MD simulation is shown in Fig.2.It is seen that the fluctuation amplitude of potential energy is small and it vibrates around-4 839kJ·mol-1,which indicates that the rPrPc system has achieved an equilibrium state.In the meantime,the RMSD of rPrPc fluctuates gently(Fig.3),which implies that one of the thermo-stable states of rPrPc has been obtained.Then the equilibrium structure of rPrPc could be used as the initial structure of SMD simulation.Both potential energy and RMSD of rPrPc show that the protein is conservative and stable during the MD simulation.This means it is hard to study the unfolding dynamics of rPrPc at short MD time,which is essential to the misfolding path of PrPc to PrPsc.

Fig.2 Potential energy of rPrPc with respect to MD simulation time
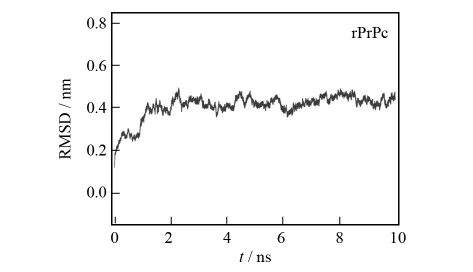
Fig.3 RMSD change of the backbone of rPrPc during the MD simulation
As shown in Fig.1,rPrPc is mainly composed of threeα-helices and one antiparallelβ-sheet.According to the residue number,they are named asα1(Asp143-Thr153),α2 (Gln171-Hse186),α3 (Glu199-Ala224),β1(Leu129-Ala132)andβ2(Gln159-Tyr161),respectively.In order to observe the flexibility of rPrPc,we separately extracted the RMSD values of the helices and sheet to compare the stability difference.As seen in Fig.4,the RMSD order is ranked asα2>α3>α1>β1>β2,which indicates that the flexibility of the helices is larger than that of the sheet.Besides,the RMSD ofα3rises at 2ns,which,with the assistance of trajectory movie,indicates that there exists an unfolding phenomenon ofα3thereat.Moreover,β1keeps twisting during the whole MD simulation,which is why the RMSD value ofβ1is lar-ger than that ofβ2(Fig.4).
In order to reveal the unfolding of the helixα3,we analyzed its relative hydrogen bond(H-bond).The H-bond distance between the oxygen atom of Gln218and the hydrogen atom of Gln222during the whole simulation is displayed in Fig.5a;and the change of another H-bond(Gln219:O-Ala223:H)distance is shown in Fig.5b.It is distinctly seen that there is distance increase of H-bond during the MD simulation.As shown in Fig.5a,the average H-bond distance is 0.22nm from 0ns to 2.4 ns;however,the average H-bond distance jumps to 0.4 nm from 2.4ns to the end of the MD simulation.Such an increase of H-bond distance enhances the unfolding ofα3in rPrPc;and the change of the H-bond distance with a large amplitude is also confirmed by the flexibility ofα-helix.
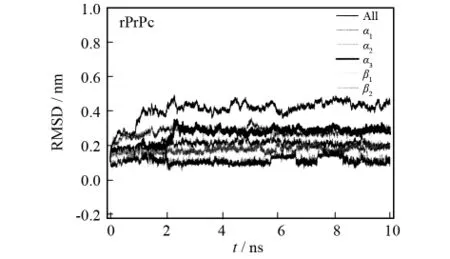
Fig.4 RMSD values of the helix and sheet structures versus MD simulation time
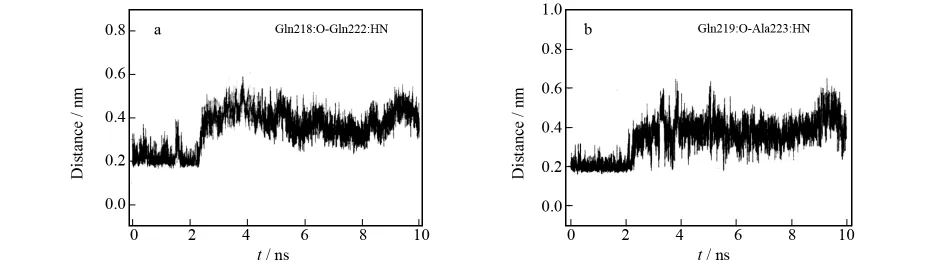
Fig.5 Relative H-bond distance ofα3in rPrPc with respect to MD simulation time
Moreover,the RMSD ofα2increases at the beginning of MD simulation(Fig.4),which is attributed to the folding ofα2in relation to thermodynamics of rPrPc.The snapshots of rPrPc at the initial state and the final state of MD simulation are shown in Fig.6aand Fig.6b,separately.The folding part ofα2is colored with red,and it is dominated by easy transformation from coil to helix.Corresponding H-bond distances(Hse186:O-Thr190:H and Val188:O-Thr192:H)shown in Fig.7also give evidences to the folding phenomenon.Both in Fig.7aand Fig.7b,the dramatic decrease of H-bond distance happens within 1ns.
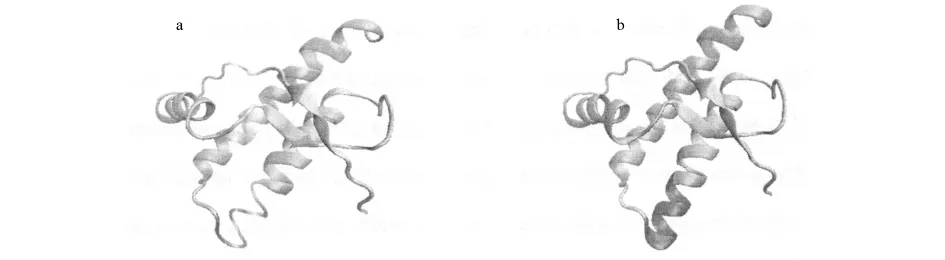
Fig.6 (a)Snapshot of the initial state of rPrPc before MD simulation and(b)Snapshot of the final state of rPrPc at the end of MD simulation
2.2 SMD simulation results
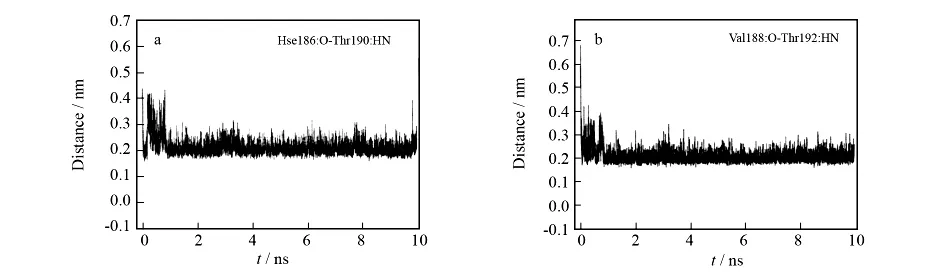
Fig.7 Relative H-bond distance ofα2in rPrPc with respect to MD simulation time
In rPrPc system,after 10ns MD simulation without applying any disturbances to the system,an equilibrium state was relegated to the SMD simulations as the starting state.During the SMD simulation,the SMD water atoms lie in the outermost layer of the water box,and the external forces used as probe tips are applied on the SMD water atoms.In PCV simulations,protein was pushed with a constant velocity from the six directions of+x,-x,+y,-y,+zand-z.The force analysis along six directions of rPrPc with respect to simulation time is shown in Fig.8.By analyzing the six force curves,we can easily find that the force endurance of rPrPc is variable in different directions.As to all the six curves in Fig.8,the fluctuation amplitude in the+yand-ydirections is obviously larger than that in the other four directions.Both the height and width of the force peak in the two directions are obviously large,which shows that the rigidity of rPrPc in these two directions is predominant.However,there are still differences between the two curves+yand-ydirections.Namely,the fluctuation of force in+ysystem is larger than that in-ysystem during the early part of SMD simulation,whereas it is opposite during the latter part of SMD simulation.From the view of SMD trajectory,we can see that SMD waters are mainly encountered helices in y-axis direction;and the three helices are staggered to form a protective wall to resist the external disturbance.Thus the external force must be enlarged to keep the SMD waters moving at a constant velocity.Since the helix wall is located at the side of+ydirection,difference of+yand-ysystems is thus caused.However,the loop structure is soft and it could be easily passed through by SMD waters.Moreover,the SMD waters could pass through the helix wall from the side with less resistance.As a result,the force applied in other directions is small.In terms of all the SMD simulations,no obvious structural deformation of rPrPc is found from the trajectory movie,which shows that the compact structure of rPrPc can efficiently remain stable under the external disturbance.
3 Conclusions
MD and SMD simulations were combined to accelerate the dynamic studies of prion system.The rPrPc was employed to reveal the stability of prion protein at atomic level.Energy minimization and MD simulation were used to get the equilibrium state of the protein,and SMD simulation was carried out to compare the rigidity distribution and dynamics characteristics of the protein in different directions.It has been confirmed that the tertiary structure of rPrPc is compact and is hard to be unfolded with thermodynamics movement.MD analysis shows that the number of dynamic forms ofα-helix is larger than that ofβ-sheet in rPrPc system.Both folding and unfolding ofα-helix appear during the simulation,and the secondary structure ofα-helix of rPrPc is more flexible thanβ-sheet.It is just the instability of the helix that might be the dynamic basis of the misfolding of PrPc into PrPsc.Besides,SMD analysis shows that the rigidity of PrPc is anisotropic and the protective wall staggered withαhelix is important in maintaining the stability of PrPc under external disturbances.However,there is no significant unfolding of helix during all the simulation processes under the selected condition.Longer time MD simulation and stronger external forces may inspire the dynamics of rPrPc,and further studies on this topic would be of significance for better understanding the pathogenesis of prion diseases.
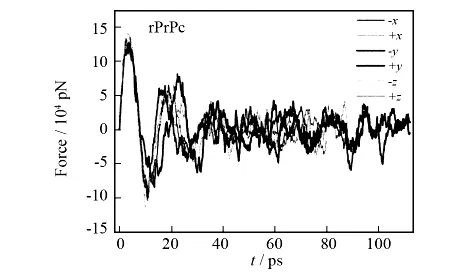
Fig.8 Force changes of rPrPc in all the six directions during the SMD simulation
[1]PRUSINER S B.Novel proteinaceous infectious particles cause scrapie[J].Science,1982,216:136-144.
[2]CAUGHEY B,CHESEBRO B.Transmissible spongiform encephathies and prion protein interconversions[J].Adv Virus Res,2001,56:277-311.
[3]AGUZZI A,POLYMENIDOU M.Mammalian prion biology:one century of evolving concepts[J].Cell,2004,116:313-327.
[4]PETSKO G A,RINGE D.Fluctuations in protein structure from X-ray diffraction[J].Annu Rev Biophys,1984,13:331-371.
[5]MARASSI F M,OPELLA S J.A solid-state NMR index of helical membrane protein structure and topology[J].J Magn Reson,2000,144:150-155.
[6]DAVID B,ANDREJ S.Protein structure prediction and structural genomics[J].Science,2001,294:93-96.
[7]PHILLIPS J C,BRAUN R,WANG Wei,et al.Scalable molecular dynamics with NAMD[J].J Comput Chem,2005,26:1781-1802.
[8]MAYORL U,GUYDOSH N R,JOHNSON C M,et al.The complete folding pathway of a protein from nanoseconds to microseconds[J].Nature,2003,421:863-867.
[9]ZHANG Jia Pu.Comparison studies of the structural stability of rabbit prion protein with human and mouse prion proteins[J].J Theor Biol,2011,269:88-95.
[10]SOTOMAYOR M,SCHULTEN K.Single-molecule experiments in vitro and in silico[J].Science,2007,316:1144-1148.
[11]KALE K,SKEEL R,BHANDARKAR M,et al.NAMD2:Greater scalability for parallel molecular dynamics[J].J Comp Phys,1999,151:283-312.
[12]MACKERELL A D,JR BASHFORD D,BELLOTT M,et al.All-atom empirical potential for molecular modeling and dynamics studies of proteins[J].J Phys Chem B,1998,102:3586-3616.
[13]WEN Yi,LI Jun,YAO Wen Ming,et al.Unique structural characteristics of the rabbit prion protein[J].J Biol Chem,2010,285:31682-31693.
[14]CHEN Xin,WU Tao,WANG Qi,et al.Shield effect of silicate on adsorption of proteins onto silicon-doped hydroxyapatite(100)surface[J].Biomaterials,2008,29:2423-2432.
Study on stability of rabbit prion protein by molecular dynamics simulation
CHEN Xin,JIN Xiang-feng,LIU Ya-fang,ZHANG Jing-lai*
(CollegeofChemistryandChemicalEngineering,HenanUniversity,Kaifeng475004,Henan,China)
Prion diseases are fatal and highly infectious neurodegenerative diseases that affect humans and animals.Rabbit is one of the several mammalian species reported to be resistant to infection from prion diseases.In this research,molecular dynamics(MD)and steered molecular dynamics(SMD)simulations were combined to study the stability of rabbit prion protein(rPrPc).The dynamics characteristics of the key secondary structures and the anisotropic resistance of external forces of rPrPc were revealed.Furthermore,the structural stability of rPrPc was confirmed with structural convergence and rigidity distribution based on the simulations.
rabbit prion protein(rPrPc);structural stability;molecular dynamics(MD)simulation;steered molecular dynamics(SMD)simulation
O643.1
A
1008-1011(2012)06-0058-06
date:2012-05-11.
This work is supported by the National Science Foundation of China(21003037and 30900236).
Biography:CHEN Xin(1981-),female,doctor,lecturer,research field:computational chemistry.*
,E-mail:zhangjinglai@henu.edu.cn.
