杜仲果实和叶片转录组数据组装及基因功能注释
2012-01-08李铁柱杜红岩刘慧敏乌云塔娜叶生晶
李铁柱,杜红岩 ,刘慧敏,乌云塔娜,王 淋,叶生晶
杜仲果实和叶片转录组数据组装及基因功能注释
李铁柱1a,杜红岩2,刘慧敏1b,1c,乌云塔娜1b,1c,王 淋1b,1c,叶生晶1b,1c
(1.中南林业科技大学a.生命科学与技术学院;b.经济林育种与栽培国家林业局重点实验室;c.林学院,湖南长沙 410004;2中国林业科学研究院经济林研究开发中心, 河南 郑州 450003)
所构建的杜仲果实和叶片转录组数据库获得了54 471 338条reads数据,包含4 902 420 420nt数据信息,对reads进行拼接,获得了452 421条Contig,总长度为90 705 736 nt; 对contig进行拼接后,获得了147 027条Scaffold,总长度为65 877 052 nt;同理,对Scaffold进一步拼接后,获得了49 610条Unigene,总长度为37 616 729 nt; Unigene和COG数据库进行比对表明,杜仲果实和叶片转录组中的Unigene根据功能大致可分为25类;杜仲果实和叶片转录组中的Unigene根据GO功能可分为生物过程、细胞组分和分子功能3大类42分支;以KEGG数据库为参考,依据代谢通路可以将转录组中的数据分成125类,包括脂类代谢,DNA剪切,植物激素生物合成,苯丙氨酸生物合成,萜类化合物与类固醇类化合物合成等。
杜仲;果实;叶片;转录组;基因功能
杜仲(Eucommia ulmoides)是一种名贵的药用植物,也是温带最具开发意义的胶原植物, 具有重要的经济价值[1-2]。常规育种与分子生物学相结合是林木遗传改良的必由之路[3]。随着后基因组时代的到来, 转录组学、蛋白质组学、代谢组学等各种组学技术相继出现, 其中转录组学是率先发展起来以及应用最广泛的技术[4-5]。转录组研究是基因功能及结构研究的基础和出发点, 了解转录组是解读基因组功能元件和揭示细胞及组织中分子组成所必需的, 并且对理解机体发育和疾病具有重要作用[6]。细胞的功能是从基因的表达开始的,转录组是指某一时间细胞内所有基因转录而来的RNA 总称[7]。通过分析转录组,可高通量地获得基因表达的 RNA 水平有关信息, 可以揭示基因表达与一些生命现象之间的内在联系。据此我们可以高通量表征细胞生理活动规律,确定细胞代谢特性,并进而对细胞进行修饰改造[8-9]。
目前关于转录组的研究也是比较多的。例如,2010年朱立煌[10]利用水稻全基因组芯片, 系统考察了超级杂交稻“两优培九”及其双亲—“培矮64s“(母本 )和“ 93—11”(父本 )在 7个不同发育时期的组织中的基因表达谱, 结果表明, 从转录谱来看, 杂种 F1与亲本间的相似性大于亲本之间的相似性; 在发现的 3 000多个杂种和亲本间差异表达的基因中, 有各种不同的差异表达类型, 多数是偏于单亲的显性表达, 但也有只在杂种中出现的超亲表达。2011年张乐等[11]以大豆基因组的46 430个高置信编码基因和2 071条大豆全长转录本序列为数据来源,应用CodonW软件对大豆全基因组密码子组成、同义密码子使用频率和全长转录组编码区密码子使用各项参数的计算和统计分析发现,基因的表达水平与编码区G+C和GC3s含量均呈极显著正相关,且G+C和GC3s含量越高的基因密码子使用偏好性越高,并确定了UCC和GCC为大豆最优密码子。2011年吴剑锋等[12]利用 tpa 及其野生型植株的开放花制备的mRNA反转录成cDNA与拟南芥ATH1芯片进行杂交,筛选出在tpa及其野生型植株中表达有差异的基因,并利用RT-PCR技术对芯片筛选出的基因进行验证,获得了152个在野生型(W1)和完全退化株(M3)转录组中差异表达的基因,61个在W1和部分退化株(I2)转录组中差异表达的基因,以及24个在I2和M3转录组中差异表达的基因,通过对41个基因的RT-PCR验证,获得了At2g42840、At1g57750、At5g20630、At2g03090、At3g08030、At5g08000、At2g28790、At5g63310 和At2g24270等9个在 tpa 及野生型植株中具显著不同的时空表达特性的基因。2010年Olivia Wilkins等[13]通过对拟南芥一天中4个不同时间点上干旱对转录组调节的影响研究支持了干旱应答转录的假说,即干旱应答转录是在一天的不同时间里,通过显著的激素和应激反应途径形成的。2006年M.Becerra等[14]通过研究生长在奶酪乳清蛋白培养基上的乳酸克鲁维酵母,与生长在合成培养基上的该酵母作比较,发现当培养基变化时,与糖基化和分泌途径相关的基因表达完全发生了重排,该酵母系统也被用来检测乳清补充剂作为抗氧化剂的优势,生长在乳清蛋白培养基上的乳酸克鲁维酵母,其和谷胱甘肽生物合成有关的基因的转录并没有增加,然而,在这种介质中生长的酵母,其和谷胱甘肽代谢和氧化应激反应有关的其他基因则出现了过量表达。
1 材料与方法
1.1 植物材料
于杜仲幼果时期(5月份左右),在国家林业局泡桐研究中心采集“华仲六号”杜仲果实和叶片为材料。
1.2 杜仲果实和叶片总RNA提取
果实和叶片RNA的提取参照陈建[16]的“几种提取杜仲RNA方法的比较”。
1.3 杜仲果实和叶片的转录组测序
转录组测序工作委托深圳华大公司完成。
1.4 杜仲果实和叶片转录组数据组装及基因功能注释技术路线

图1 数字化转录组数据库的分析Fig. 1 Data Analysis of Digital Transcriptome
本研究对杜仲果实和叶片合成调控时期的转录组进行测序,展开了对数据库中Unigene的全面分析和注释。具体流程如图1所示。
2 结果与分析
对杜仲果实和叶片进行测序后,共获得了54 471 338个reads片段,包含了4 902 420 420个核苷酸序列信息,其中片段长度大于20个碱基的百分比为96.05%,中间未知序列的片段为0,GC%值为47.00%,由此可以看出此次转录组测序结果较好,可为后续的数据组装提供很好的原始数据。
2.1 杜仲果实和叶片转录组数据的组装
对13 333 334个reads片段采用over-lap的方法进行拼接,共获得了452 421个Contig片段,序列信息达到了90 705 736 nt;其中,长度在75~100 nt范围内的Contig片段有273 533条,比
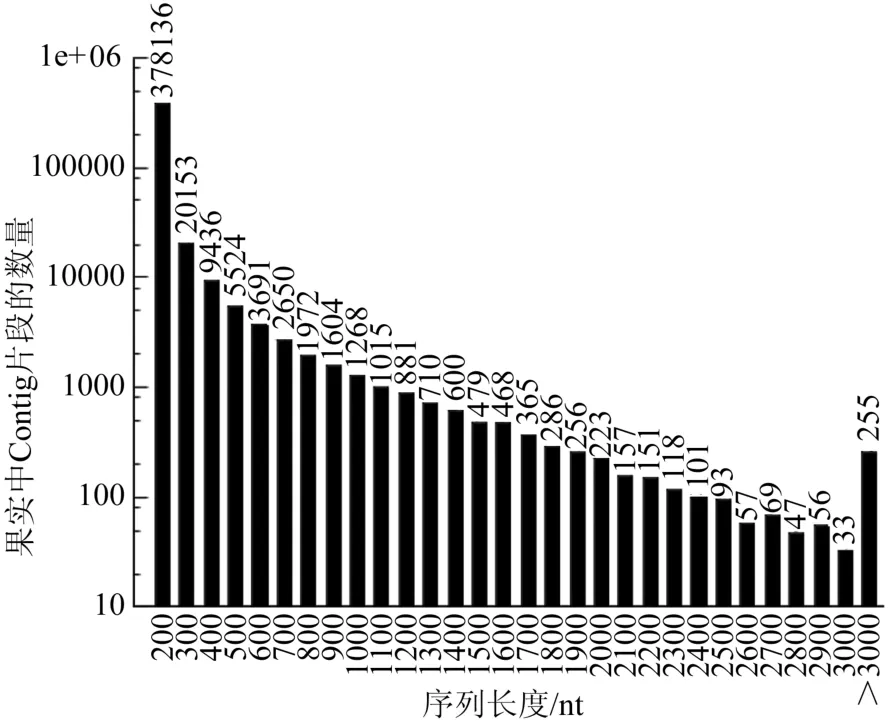
图2 杜仲果实转录组的Contig数据长度分布图Fig. 2 Contig Data Length Distribution for Digital Transcriptome of Eucommia ulmoides Oliv Fruits
在Contig数据的基础上,进一步采用over-lap的方法进行拼接,共获得了147 027个Scaffold片段,序列信息达到了65 877 052 nt;其中,长度在100~500 nt范围内的Scaffold片段有111 874条,比例达到了76.09%;500~1 000 nt的有19 303条,比例为13.13%;1 000~1 500 nt的有7 730条,占5.26%;1 500~2 000 nt的有4 113条,占2.80%;≥2 000 nt的有4 007条,2.73%(见表2、图4、图5)。
在Scaffold数据的基础上,进一步拼接,数据共获得了49 610个Unigene片段,序列信息达到了37 616 729 nt,片段大小从100~3 256 nt;其中,长度在100~500 nt范围内的Unigene片段有27 428条,比例达到了55.29%;500~1 000 nt的有例达到了60.46%;100~200 nt的有80 470条,比例为17.79%;而≥200 nt的共有88 087条,比例为19.47%(见表1、图2、图3)。由此可见,Contig数据主要以75~100 nt的为主,完全符合Illumina测序的预期结果。10 240条,比例为20.64%;1 000~1 500 nt的有5 136条,占10.35%;1 500~2 000 nt的有3 215条,占6.48%;≥2 000 nt的有3 591条,占7.24%(见表3、图6、图7)。

表1 杜仲果实和叶片转录组的Contig数据组装质量统计Table 1 Data Assembly for Contig in Digital Transcriptome of Eucommia ulmoides Oliv Fruits and Leaves

图3 杜仲叶片转录组的Contig数据长度分布图Fig. 3 Contig Data Length Distribution for Digital Transcriptome of Eucommia ulmoides Oliv Leaves
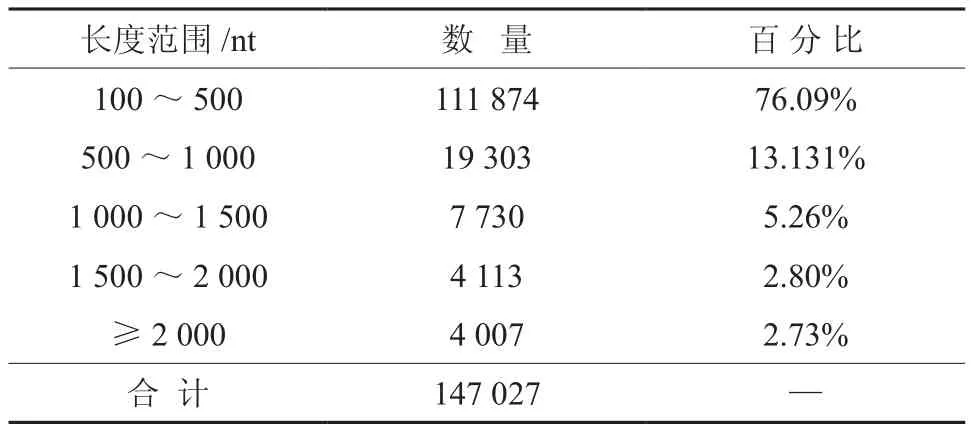
表2 杜仲果实和叶片转录组的Scaffold数据组装质量统计Table 2 Data Assembly for Scaffold in Digital Transcriptome of Eucommia ulmoides Oliv Fruits and Leaves
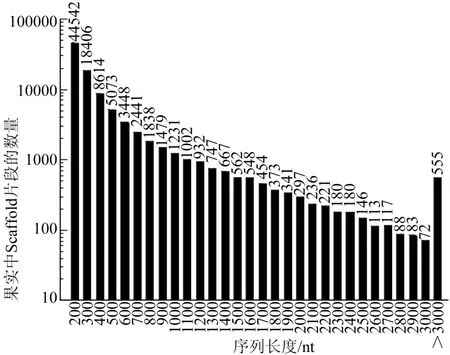
图4 杜仲果实转录组的Scaffold数据长度分布图Fig. 4 Scaffold Data Length Distribution for Digital Transcriptome of Eucommia ulmoides Oliv Fruits

图5 杜仲叶片转录组的Scaffold数据长度分布图Fig. 5 Scaffold Data Length Distribution for Digital Transcriptome of Eucommia ulmoides Oliv Leaves

表3 杜仲果实和叶片转录组的Unigene数据组装质量统计Table 3 Data Assembly for Unigene in Digital Transcriptome of Eucommia ulmoides Oliv Fruits and Leaves
对Unigene进行覆盖度的分析发现,94 334条Unigene能够与测序的原始数据reads相对应,且不同的Unigene与reads的对应关系各不相同,分别用测序深度—depth(reads碱基数/ 序列长度)和覆盖度—coverage( 中有reads覆盖的碱基数/ 序列长度)表示,depth的范围在0.038 3~17 784.1之间,coverage的范围在3.83%~100%之间;所有的43 879条没有出现未知序列,只有2 046条有未知序列;GC%为22.07%~71.09%;Unigene对应的reads,即样品中能唯一比对到指定Unigene序列的reads数(Unique-mapped-Reads)从1~81 612条范围不等。
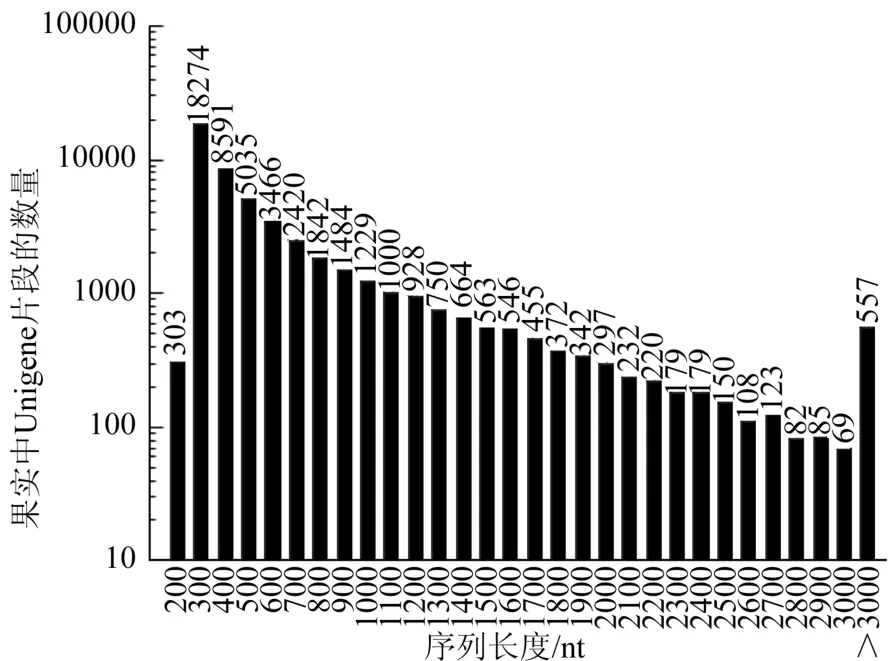
图6 杜仲果实转录组的Unigene数据长度分布图Fig. 6 Unigene Data Length Distribution for Digital Transcriptome of Eucommia ulmoides Oliv Fruits
2. 2 杜仲转录组Unigene的cds的blast分析
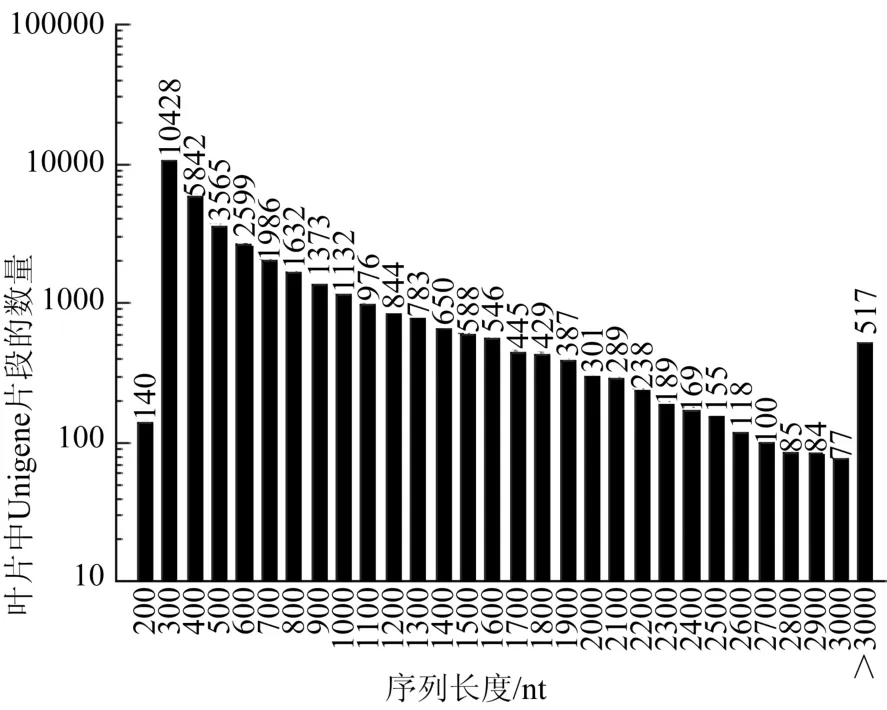
图7 杜仲叶片转录组的Unigene数据长度分布图Fig. 7 Unigene Data Length Distribution for Digital Transcriptome of Eucommia ulmoides Oliv Leaves
对杜仲转录组所有的Unigene的cds进行blast分析,共发现了30 280个Unigene片段,片段大小从100~3 256 nt;其中,长度在100~~500 nt范围内的Unigene片段有15 138条,比例达到了50.00%;500~1 000 nt的有9 913条,比例为32.74%;1 000~1 500 nt的有5 204条,占 17.19%;1 500~ 2 000 nt的 有2606条, 占8.61%;≥2 000 nt的有2009条,占6.63%(见表4、图8)。
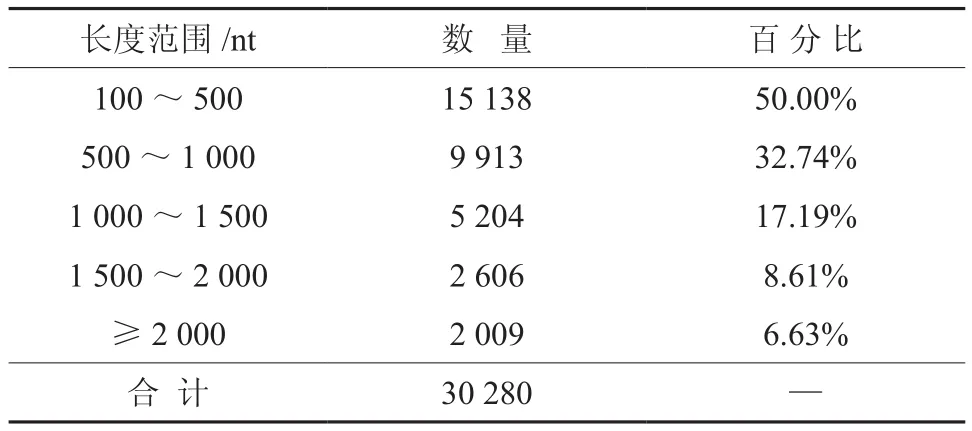
表4 杜仲转录组All-Unigene的cds的blast分析表Table 4 Eucommia transcriptome All-Unigene cds blast analysis
在蛋白质数据库中对杜仲转录组所有的Unigene进行blast分析后,共发现了28 074个Unigene片段,片段大小从100~3 256 nt;其中,长度在100~500 nt范围内的Unigene片段有26 575条,比例达到了94.66%;500~1 000 nt的有5 268条,比例为18.76%;1 000~1 500 nt的有588条,占2.09%;1 500~2 000 nt的有92条,占0.33%;≥2 000 nt的有31条,占0.11%(见表5、图9)。
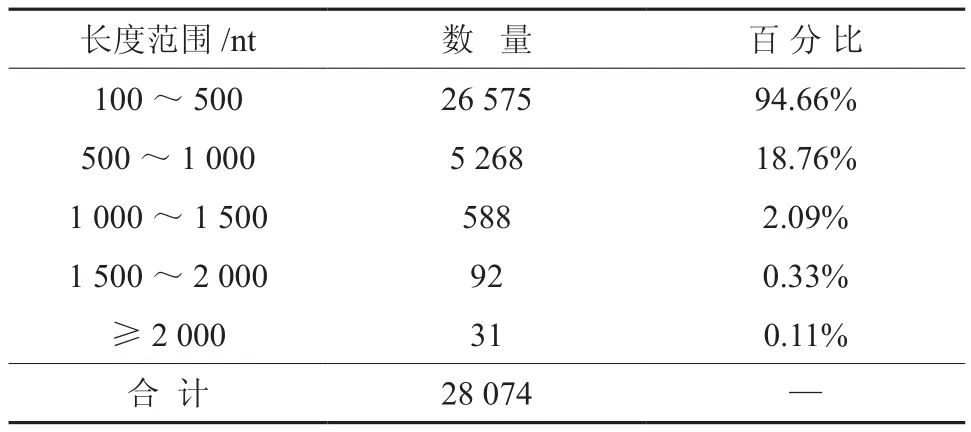
表5 杜仲转录组All-Unigene在蛋白质数据库中的blast分析Table 5 Eucommia transcriptome All-Unigene blast in the protein database

图8 杜仲转录组All-Unigene的cds的blast分析图Fig.8 Eucommia transcriptome All-Unigene cds blast analysis
在ESTscan数据库中对杜仲转录组的cds进行分析,共发现1 595个Unigene片段,片段大小从100~2 000 nt;其中,长度在100~500nt范围内的Unigene片段有1394条,比例达到了87.40%;500~1 000 nt的有180条,比例为11.29%;1 000~1 500 nt的有16条,占1.00%;1 500~2 000 nt的有92条,占0.19%;≥2 000 nt的有31条,占0.06%(见表6、图10)。
表6 杜仲转录组All-Unigene在ESTscan数据库中的cds分析Table 6 Eucommia transcriptome All-Unigene analysis in ESTscan the database cds

图9 杜仲转录组All-Unigene在蛋白质数据库中的blast分析图Fig.9 Eucommia transcriptome All-Unigene database of protein blast analysis
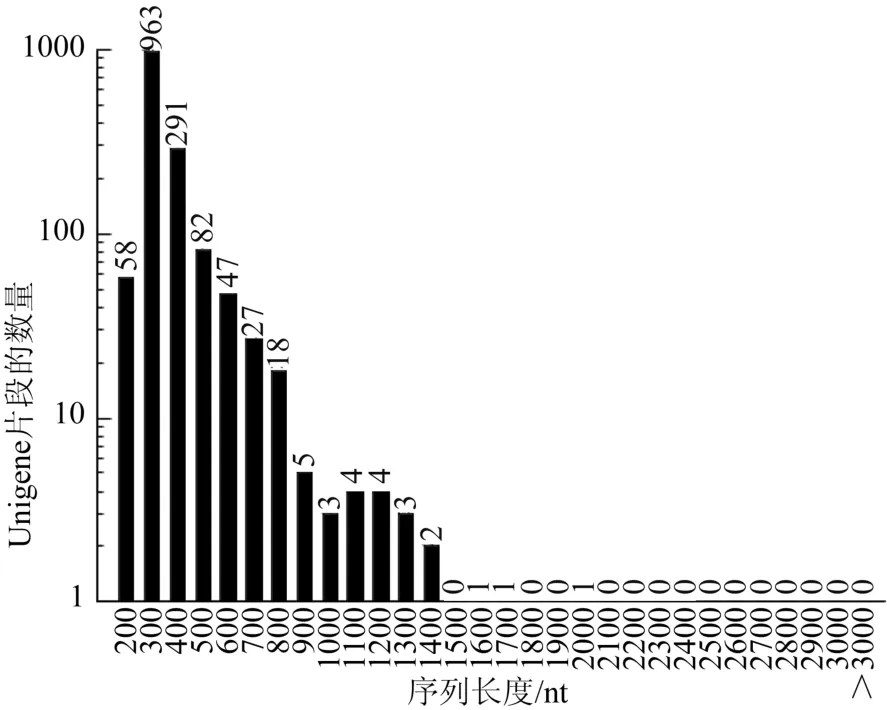
图10 杜仲转录组All-Unigene在ESTscan数据库中的cds分析图Fig.10 The cds Eucommia transcription group All-Unigene ESTscan database analysis
2.3 杜仲转录组Unigene的COG功能分类
共有125 934条 可以与数据库中的基因具有相似性,且较多的单条Unigene能够与多种基因相对应,建立了125 934条对应关系。杜仲果实和叶片转录组中的Unigene根据功能大致可分为25类(图11中用A~Z表示),并对每一类的基因数量进行了统计(表7)。从表中和图中可以看出,Unigene的COG功能种类比较全面,涉及了大多数的生命活动,整体功能类的基因数量最多,有20 153条;核结构相关基因类的数量最少,只有3条;其他种类基因的表达丰度不尽相同,具体种类和数量见表7。

表7 杜仲果实和叶片转录组的UnigeneCOG功能分类Table 7 COG Function Classification of Eucommia ulmoides Oliv Fruits and Leaves in Digital Transcriptome
2. 4 Unigene的GO功能分类分析
根据分析发现,共有8 260条Unigene可以与数据库中的基因具有相似性,且较多的单条Unigene能够与多种基因相对应,建立了8 260条对应关系,从而得到尽可能多的注释和分类。杜仲转录组中的Unigene根据GO功能大致可分为生物过程、细胞组分和分子功能3大类42分支(见图12),并对每一类的基因数量进行了统计(见表8)。从表中和图中可以看出,其中细胞组分这一大类中,涉及的基因最多,有3 959条;在生物过程这一大类中,涉及的基因有2 602条;在分子功能这一大类中,涉及的基因有2 665条。其他种类基因的表达丰度不尽相同,具体种类和数量见表8。

表8 杜仲果实和叶片转录组的UnigeneGO功能分类Table 8 Gene Ontology of Eucommia ulmoides Oliv Fruits and Leaves Unigene in Digital Transcriptome
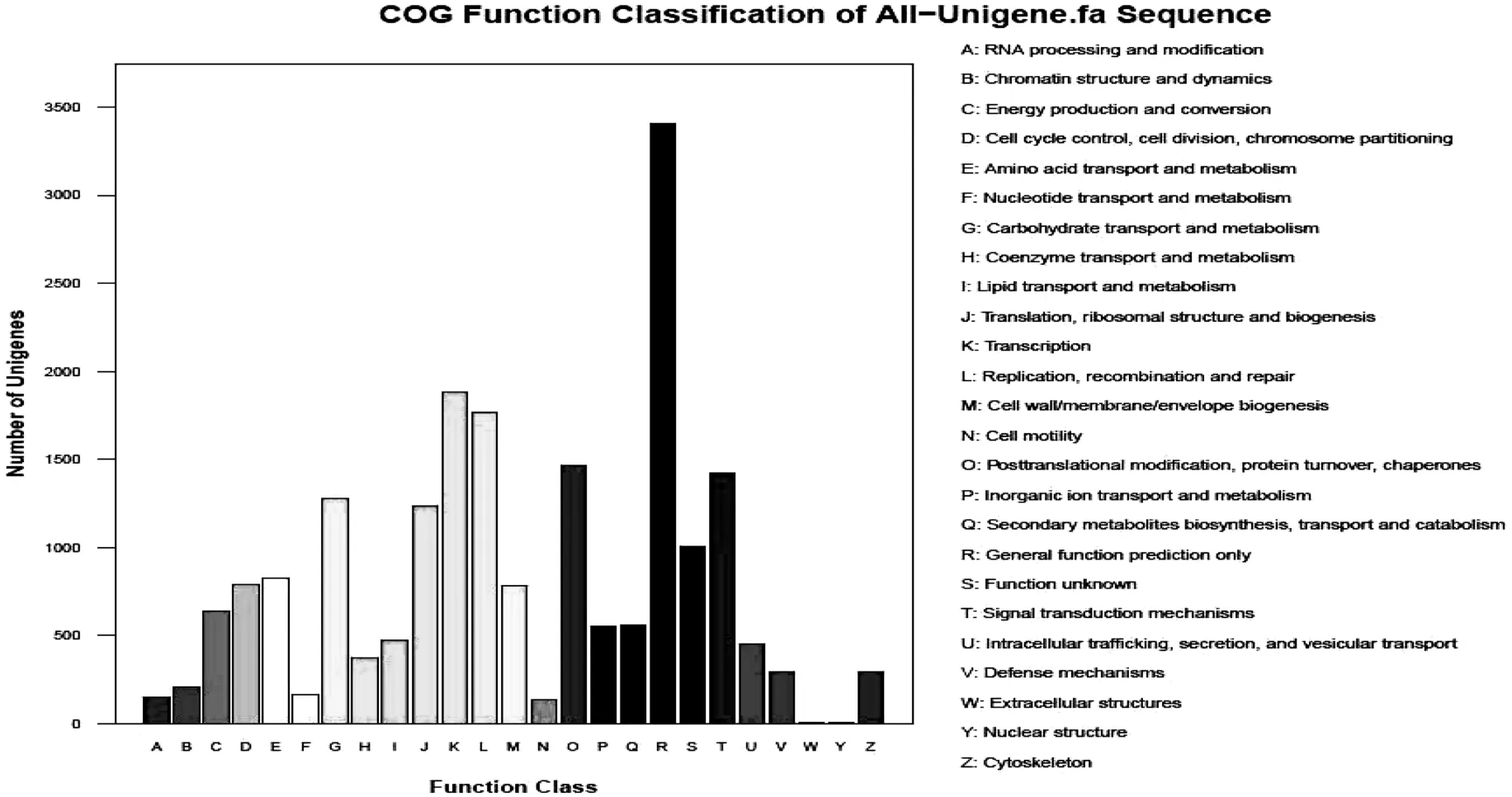
图11 杜仲果实和叶片转录组的UnigeneCOG功能分类Fig. 11 COG Function Classification of Eucommia ulmoides Oliv Fruits and Leaves in Digital Transcriptome

图12 杜仲果实和叶片转录组的UnigeneGO功能分类Fig. 12 Gene Ontology of Eucommia ulmoides Oliv Fruits and Leaves Unigene in Digital Transcriptome
2.5 Unigene 的KEGG代谢途径分类
利用KEGG数据库作为参考,依据代谢通路可以将转录组中的数据分成125类,包括生化代谢通路,植物—真菌互作,DNA剪切,植物激素生物合成,苯丙氨酸生物合成,萜类化合物与类固醇类化合物合成,脂类代谢,RNA降解等,具体列于表9。其中,不饱和脂肪酸的生物合成,涉及的基因有93条,占整体的0.63%;黄酮类代谢,涉及的基因有249条,占整体的1.68%;苯丙素类代谢,涉及的基因有421条,占整体的2.84%;类胡萝卜素类代谢,涉及的基因96条,占整体的0.61%;芸苔素类代谢,涉及的基因25条,占整体的0.17%;叶酸代谢途径,涉及的基因13条,占整体的0.09%;维生素B5代谢途径,涉及的基因31条,占整体的0.21%;维生素B2代谢途径,涉及的基因有24条,占整体的0.16%;α-亚麻酸代谢途径,涉及的基因有138条,占整体的0.93%。
3 结论与讨论
所构建的杜仲果实和叶片转录组数据库获得了54 471 338条reads数据,包含4 902 420 420 nt数据信息,对reads进行拼接,获得了452 421条Contig,总长度为90 705 736 nt; 对contig进行拼接后,获得了147 027条Scaffold,总长度为65 877 052 nt;同理,对Scaffold进一步拼接后,获得了49 610条Unigene,总长度为37 616 729 nt;对杜仲转录组所有的Unigene的cds进行blast分析,共发现了30 280个Unigene片段, 在蛋白质数据库中对杜仲转录组所有的Unigene进行blast分析后,共发现了28 074个Unigene片段,在ESTscan数据库中对杜仲转录组的cds进行分析,共发现1 595个Unigene片段,共有125 934条 Unigene可以与COG数据库中的基因具有相似性,共有8 260条Unigene可以与GO数据库中的基因具有相似性。
2010年高珍[16]通过对浒苔测序得到29 370 732条reads片段,平均长度75 bp,总长为2 202 804 900 bp,将Reads数据组装成contig,得到2 288 272条contig片段,平均长度为40 bp,总长91 817 153 bp。
2011年LIU Xin- xing等[17]通过对四倍体拟南芥的转录组的研究,得到了23 476 310条reads片段,碱基对总长度达到了1 690 294 320 bp,非冗余副本(≥100 bp)的数量为125 953条,转录的中长度为41 665 175,经过组装,得到的contigs(≥100 bp)的数量从155 306( kmer =33 bp)条到23 117 ( kmer = 57 bp)条不等。
将本次测序结果与上述两人的测序结果相比,可以看出本次测序得到的数据量更大,所以本次测序结果较好。
[1] 杜红岩,李 钦,杜兰英,等.杜仲雄花茶营养成分的测定分析[J]. 中南林业科技大学学报,2007,27(6):88-89.
[2] 薛 萍.杜仲研究现状与发展前景[J].经济林研究,1995,13(3).
[3] 陈 英,江香梅,张 露,等.基于油茶59万条EST序列的转录组学初步分析[J].林业科学,2011,47(2):161-163.
[4] Lockhart DJ, Winzeler EA. Genomics, gene expressand DNA arrays. Nature, 2000, 405(6788): 827–836.
[5] 房学爽,徐刚标.表达序列标签技术及其应用[J].经济林研究,2008,26(2):127-130.
[6] 祁云霞,刘永斌,荣威恒. 转录组研究新技术:RNA-Seq 及其应用[J].遗传, 2011,33(11): 1191-1202.
[7] 井赵斌,魏 琳,俞 靓,等.转录组测序及其在牧草基因资源发掘中的应用前景[J].草业科学,2011,28(7): 1364-1369
[8] Jewett MC, Oliveira AP, Patil KR, et al. The role of highthroughput transcriptome analysis in metabolic engineering.Biotechnol Bioproc Eng, 2005, 10: 385-399.
[9] Donson J, Fang Y, Espiritu-Santo G, et al. Comprehensive gene expression analysis by transcript profiling. Plant Mol Biol, 2002,48: 75-97.
[10] 朱立煌.超级杂交水稻LYP9及其亲本的转录组学研究[J].中国基础科学·研究进展.
[11] 张 乐,金龙国,罗 玲,等. 大豆基因组和转录组的核基因密码子使用偏好性分析[J]. 作物学报, 2011,37(6):965-974.
[12] 吴剑锋,张海娟,卢海宇,等. 基因芯片分析芜菁雌蕊退化突变体 tpa 及野生型开放花的转录组差异[J]. 中国农业科学 ,2011,44(5):972-981.
[13] Olivia Wilkins, Katharina Brautigam, Malcolm M.Campbell.Time of day shapes Arabidopsis drought transcriptomes[J]. The Plant Journal,2010( 63):715-727.
[14] M Becerra, M I Gonzalez-Siso, M E Cerdan. A transcriptome analysis of Kluyveromyces lactis growing in cheese whey[J].International Dairy Journal , 2006 (16):207-214.
[15] 陈 建.几种提取RNA方法的比较[J].林业科技开发,2007,21(5):19-21.
[16] 高 珍.浒苔生理生态特性和转录组研究[D].甘肃:甘肃农业大学,2010.
[17] LIU Xin- xing, CHEN Chao. De Novo Assembly of Allotetraploid Arabidopsis suecica Transcriptome using Short Reads for Gene Discovery and Marker Identification[J]. 中国生物工程杂志, 2011,31(7): 45-53.
Transcriptome data assembly and gene function annotation of Eucommia fruits and leaves
LI Tie-zhu1a, DU Hong-yan2, LIU Hui-min1b,1c, WUYUN Ta-na1b,1c, WANG Lin1b,1c,YE Sheng-jing1b,1c
(1a. School of Life Science&Technology; 1b. Key Lab of Non-wood Forest Product of Forestry Ministry; 1c.School of Forestry, Central South University of Forestry and Technology, Changsha 410004, Hunan, China; 2.Non - timber Forestry Research and Development Center, CAF, Zhengzhou 450003, Henan, China)
The transcriptome library of Eucommia fruits and leaves contained 54471338 reads consist of 4902420420 nt. 452421 Contigs were obtained by assembling the reads, and the total length of all Contigs was 90705736 nt;In the transcriptome library, 147027 Scaffolds were obtained by assembling the Contigs, and the total length of all Scaffolds was 6587705 nt; Also the 49610 Unigenes were obtained by by assembling the Scaffolds in the transcriptome library, and the total length of all Unigenes was 37616729 nt;Unigene and the COG database to compare, Unigene in the transcriptome of Eucommia fruits and leaves can be broadly divided into 25 classes according to the function; The Unigenes GO functions in the the transcriptome library were classificated into 3 categories: biological process, cellular component and molecular function and 42 branches;In this study,the KEGG database as a reference,data in the transcriptome can be divided into 119 classes,according to the metabolic pathway,such as lipid metabolism, DNA replication, biosynthesis of plant hormones,Biosynthesis of phenylpropanoids, Biosynthesis of terpenoids and steroids and so on.
Eucommia ulmoides;fruits;leaves; transcriptome;Gene Ontology
S722.3;Q945
A
1673-923X(2012)11-0122-09
2012-10-10
国家林业公益性行业科研专项(201004029);国家十二五科技支撑计划(2012BAD21B0502)
李铁柱(1979-),男,河南南阳人,博士研究生,主要从事生态学研究
杜红岩(1963-),男,河南中牟人,研究员,博士生导师,主要从事杜仲育种栽培与综合利用的研究;E-mail:dhy515@126.com
[本文编校:欧阳钦]
