干燥和压榨作用下纤维素水解障碍机理
2012-01-05杨晓敏万金泉马邕文
杨晓敏 万金泉,2 马邕文,2 王 艳
(1.华南理工大学环境科学与工程学院,广东广州,510640;2.华南理工大学制浆造纸工程国家重点实验室,广东广州,510640)
随着世界性能源危机的加剧,寻找替代石油、煤炭等化石燃料的新能源是科研工作者的当务之急。作为一种储量丰富的可再生能源,纤维素的水解利用引人注目,一般采用的原料是秸秆、稻草等原生植物纤维,而再生植物纤维乙醇化的报道很少。随着植物资源日益短缺和对环境保护的重视,再生植物纤维的回收利用以及将其用于水解已成为一种发展趋势,然而如何将其有效转化成为葡萄糖还存在很多障碍。
近年来,关于纤维素大分子的聚集态结构的研究主要集中在纤维素各种结晶变体间的转化,广角X射线衍射(XRD)和红外光谱(FT-IR)是研究纤维素结构的有效手段。其中,XRD在确定纤维素晶型转化、纤维素结晶度、晶面指数和晶面尺寸等方面发挥了重要作用[1-3]。FT-IR是研究纤维素分子链中氢键结构的有效方法[4-5],氢键的形成使纤维素分子链的网络结构更为牢固,不利于纤维素的化学改性[6-8]。将XRD和FT-IR结合研究氢键类型间的转化可以作为研究各种工艺条件下纤维素晶体结构变化的一种新思路[9-10]。纤维素的氢键包括分子间氢键和分子内氢键,氢键类型和其相对含量的分布影响纤维素晶型、改性等物理化学性能。本研究通过改变不同的干燥和压榨条件,结合XRD和FT-IR,从纤维素晶型变化以及氢键模式分布出发,研究其对再生植物纤维纤维素催化水解过程的影响,为进一步研究纤维结构,提高植物纤维纤维素水解率奠定基础。
1 实验
1.1 原料
初始原料为桉木,经硫酸盐法蒸煮(蒸煮条件:用碱量17%,硫化度25%,液比1∶4;用碱量和硫化度均以Na2O计)脱除木素,晒干后备用。
采用GBJ-A标准疏解器将脱除木素的纤维疏解后在Frank Rapid Kothen抄片器上按GB7981—87标准抄成定量为60 g/m2的手抄片。在抄纸过程中:压榨分别采用0.1、0.2、0.3、0.4 MPa 进行2 次压榨,正反面各1次,然后自然风干;分别采用60、80、100、120℃对样品进行干燥,干燥时间为15 min。所得手抄片使用微型植物粉碎机粉碎后作为水解反应原料备用。
固体酸催化剂的制备:称取20 g微晶纤维素在400℃ N2气氛下加热6 h。所得黄黑色固体经过研细后,取1 g加入10 mL浓硫酸,在150℃ N2气氛下处理10 h。随后冷却到室温,用80℃以上的热水洗涤,直至洗液中检测不出SO2-4为止,即得到相应的磺化碳固体酸催化剂。
1.2 实验方法
催化纤维水解反应在25 mL玻璃反应管中进行,反应开始前将催化剂、再生植物纤维和蒸馏水按照一定比例加入,通过油浴锅控制反应温度和磁力搅拌速率。反应结束后取出玻璃反应管自然冷却,反应液经0.22 μm滤膜过滤,转移至试管中待测定。

表1 HPAEC-PAD的测试条件
1.3 分析方法
葡萄糖采用HPAEC-PAD(Dionex ICS-3000)进行分析,测试条件见表1。
XRD测定采用德国 Bruker公司产的 D8 ADVANCE型X射线衍射仪。测试条件:Cu靶,Lynx-Exe阵列探测器,管压40 kV,管流40 mA,扫描步长 0.04°,扫描速率0.2 s/步,扫描范围 4°~60°。
FT-TR测定采用美国Nicolet公司生产的NEXUS 670型傅里叶变换红外光谱仪,OMNIC7.3.0.94操作软件,扫描范围4000~500 cm-1。
TOC测定采用日本岛津TOC-4100总有机碳分析仪测定。TOC为水解液中的碳含量,是由固相转化而来,其变化可以间接衡量纤维的水解程度。
2 结果与讨论
2.1 干燥温度对再生植物纤维纤维素的影响
2.1.1 样品的XRD谱图分析
图1为不同干燥温度下再生植物纤维纤维素的XRD谱图。从图1可以看出,不同样品特征衍射峰的位置基本没变,说明晶型没有发生改变,仍然是纤维素Ⅰ,但是随干燥温度的升高,样品的衍射峰相对强度明显增强,特别是002面的结晶峰,其相对衍射强度分别为1660、2391、3000、3289。

图1 不同干燥温度下再生植物纤维纤维素的XRD谱图
2.1.2 纤维素的结晶度和晶面尺寸的变化
对于不同干燥温度处理后植物纤维纤维素的XRD谱图进行分峰拟合分析,分峰后得到的不同干燥温度下纤维素的结晶度和晶面尺寸的参数列于表2。由表2可以看出,随干燥温度的升高,结晶度明显 上 升, 分 别 为 57.41%、64.37%、68.10% 和68.93%,同时垂直002面微晶体尺寸也发生改变,4种样品的晶面尺寸经过计算分别为11.38、11.87、13.10、12.64 nm,微晶横截面积分别为 139.18、151.88、164.08、159.26 nm2。由此可以看出,在低于100℃时,随着干燥温度的升高,晶面尺寸与横截面积明显增大,与结晶度的变化规律一致。但经过120℃处理后,结晶度仍然增大,晶面尺寸与横截面积却减小。这可能是由于纤维素分子链表面的羟基与水分子结合的氢键较弱,在干燥过程中发生脱水断裂,断裂后的自由羟基之间重新结合形成新的分子内和分子间氢键,使纤维素分子链排列更加有序,从而导致结晶度上升。当温度升高至120℃,新形成的分子间氢键在热力作用下收缩,平行的晶面之间聚集使得晶面尺寸减小,导致纤维素的共结晶现象[11-12]。
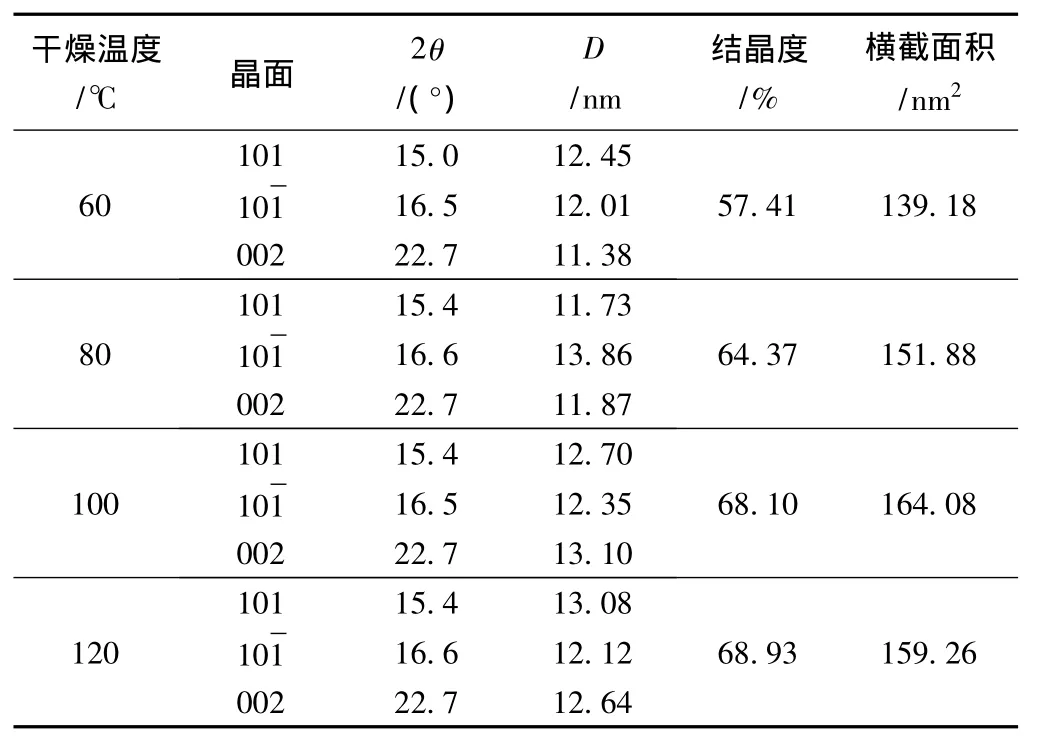
表2 不同干燥温度下纤维素结晶度和晶面尺寸的变化
2.1.3 FT-IR分析
图2为不同干燥温度下再生植物纤维纤维素的红外谱图。如图2所示,经不同温度处理后,没有新的吸收峰出现,说明没有在纤维素分子中引入新的基团;吸收峰强度的差异表明干燥温度对原有基团的振动强度产生了影响。随干燥温度的升高,代表氢键的3650~3000 cm-1间的吸收峰强度有所降低,这主要是因为经干燥后,胞腔发生塌陷,形成部分不可逆的键合。而120℃时氢键吸收峰明显增强,可能是因为温度的提高使得纤维素分子链的断面运动加剧,氢键重排的可能性变大,这一点也与采用XRD分析的结论一致。
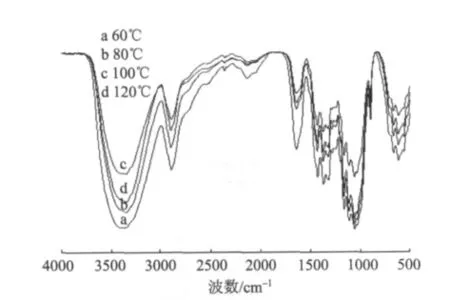
图2 不同干燥温度下再生植物纤维纤维素的红外谱图
2.1.4 红外谱图的氢键拟合分析
纤维素结晶区的羟基通过氢键产生作用,由于氢键分为分子内氢键和分子间氢键,表现在红外光谱的吸收波数是不一样的。对红外谱图求二阶导数,寻找分峰的位置,根据文献报道[13],纤维素Ⅰ分子内氢键O(2)H…O(6)、O(3)H…O(5)和分子间氢键O(6)H…O(3')的特征吸收波数分别为3455~3410、3375~3340、3310~3230 cm-1,由此结合导数光谱进行高斯分峰拟合。
图3列出了不同干燥温度下纤维素的红外谱图氢键区域拟合,分出了纤维素中的3种主要氢键形式,分别用谱带1、2、3表示。表3列出了氢键拟合结果和分峰的强度。
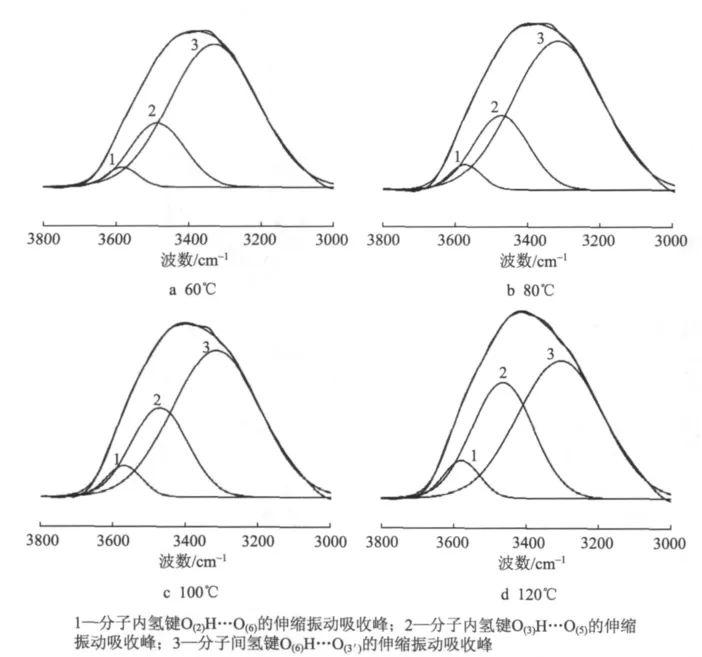
图3 不同干燥温度下纤维素红外谱图氢键区域拟合图
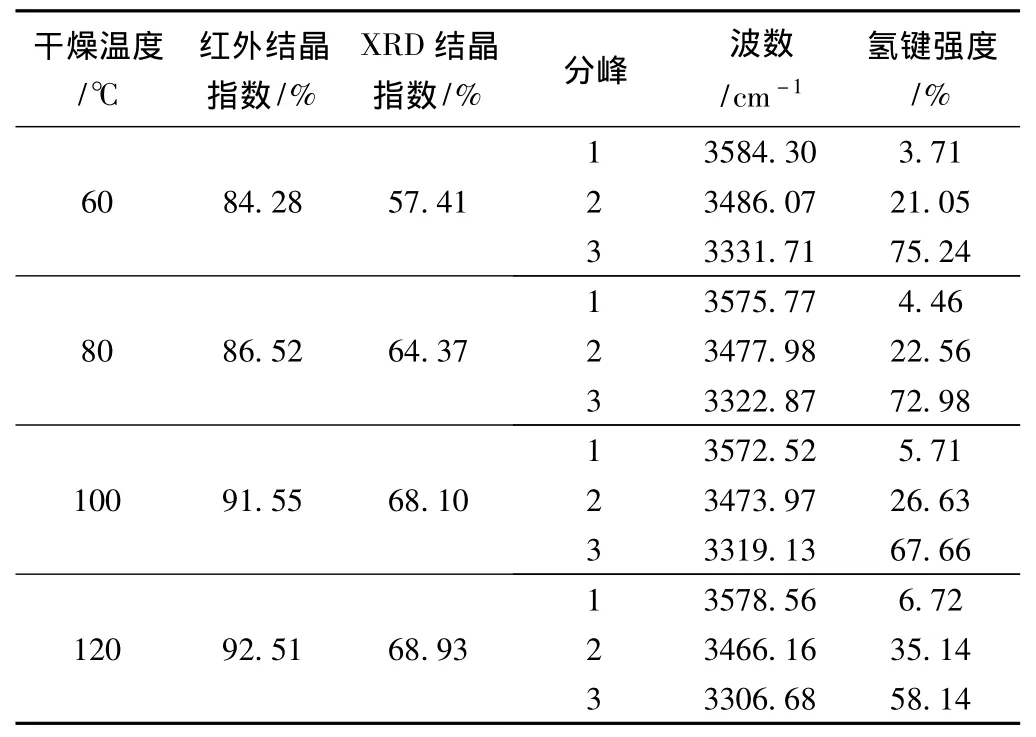
表3 不同干燥温度下纤维素红外谱图氢键区域的拟合结果
从4个样品的红外谱图氢键区域拟合结果可以看出,再生植物纤维纤维素中分子间氢键占据主导,纤维素链间主要靠分子间氢键结合在一起,稳定纤维素链,而分子内氢键处于辅助地位,符合分子间氢键的键能大于分子内氢键的规律[14]。随干燥温度的升高,样品结晶度增大,4个样品的分子内氢键O(2)H…O(6)的强度从3.71%上升到6.72%,分子内氢键O(3)H…O(5)的强度从21.05%上升到35.14%,分子间氢键O(6)H…O(3')的强度从75.24%下降到58.14%。这主要是由于随干燥温度的升高,纤维素分子中的结合水被不断脱除,纤维素表面的自由羟基会相互结合形成新的分子内和分子间氢键,当温度超过100℃时,纤维素分子内氢键的强度明显增大,从而使无定形区有序性增强,纤维内部晶态结构更稳定,导致纤维难于水解。
2.1.5 再生植物纤维纤维素催化水解产糖率分析
纤维素微观结构的变化必然会对其宏观性质和反应性能造成影响。如图4所示,在干燥温度为60℃时,纤维素水解率和葡萄糖产率最高,之后随干燥温度的升高,纤维素水解率逐渐降低,葡萄糖产率也随之降低,两者的变化与纤维素结晶度的变化规律一致,由此可以看出,干燥温度通过对纤维素结晶结构等微观结构的影响进一步影响其水解性能。在催化水解过程中,温度的升高使得纤维素的无定形区更有序,阻碍了催化剂的有效官能团与纤维素自由羟基之间的接触,从而增大了纤维素水解的难度,导致葡萄糖产率下降。
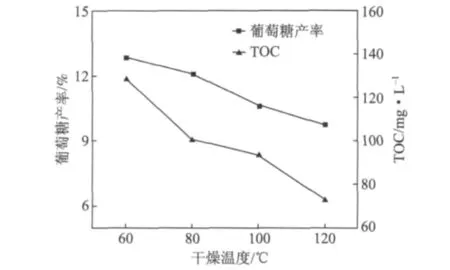
图4 不同干燥温度下再生植物纤维纤维素催化水解产糖率的变化
2.2 压榨压力对再生植物纤维纤维素的影响
2.2.1 样品的XRD谱图分析
图5为不同压力条件下再生植物纤维纤维素的XRD谱图。从图5可知,不同样品特征衍射峰的位置基本没变,说明晶体结构没有发生改变,仍然是典型的纤维素Ⅰ,但是随压榨压力的升高,样品的衍射峰相对强度逐渐增强,002面的结晶峰相对衍射强度分别为1986、2857、3142、3581。
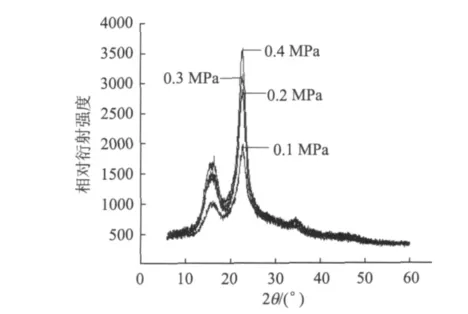
图5 不同压力条件下再生植物纤维纤维素的XRD谱图
2.2.2 纤维素的结晶度和晶面尺寸的变化
对于不同压榨压力的纤维素XRD谱图进行分峰拟合分析,分峰后得到了不同压榨压力下再生植物纤维纤维素的结晶度和晶面尺寸的参数(见表4)。随压榨压力的增大,结晶度明显上升,分别为53.58%、65.14%、67.92% 和 70.09%,同时垂直002面微晶体尺寸也随之增大,4种纤维素样品的晶面尺寸分别为 11.60、12.83、14.34、17.11 nm,微晶横截面积分别为215.18、218.05、275.11、286.76 nm2。由此可以看出,经压力作用后,纤维素的结晶度以及微晶体尺寸都明显增大,这可能是纤维素各晶区之间在压力作用下相互靠近发生了晶区间的合并所致,同时压力的增大使得纤维素分子链被压实,分子间更易结合,特别是无定形区内部的自由羟基得以靠近并结合,从而导致结晶度上升。
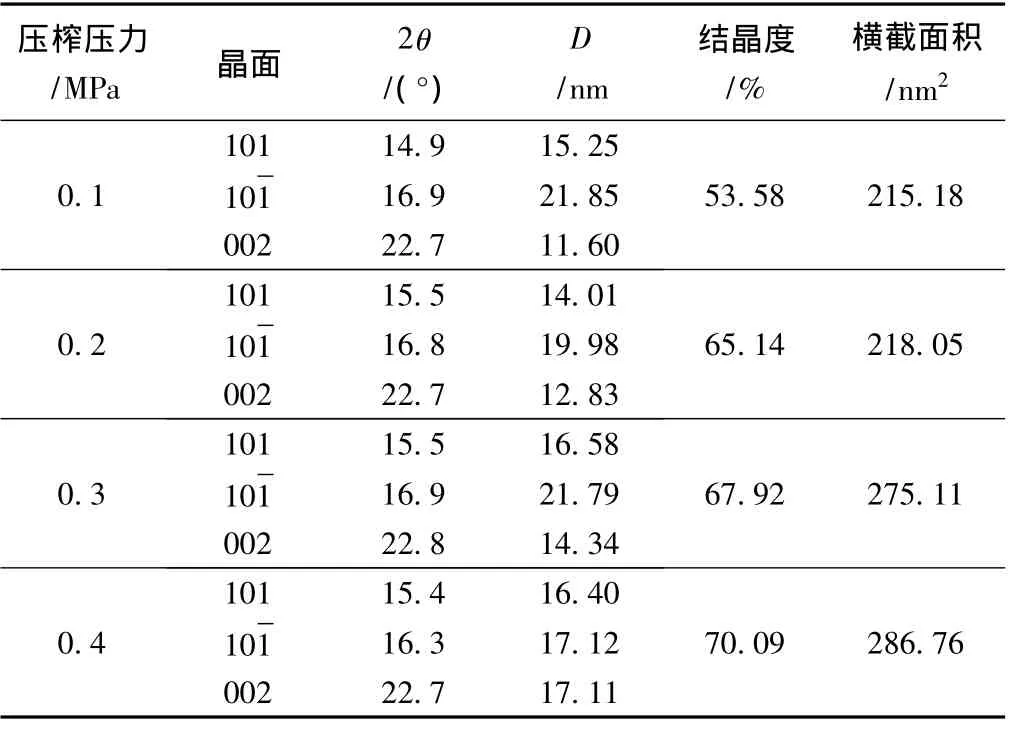
表4 不同压榨压力下纤维素结晶度和晶面尺寸的变化
2.2.3 FT-IR分析
图6为不同压榨压力条件下再生植物纤维纤维素的红外谱图。从图6可知,4个样品的红外谱图基本相同,经过压力作用后,没有新的吸收峰出现,说明在纤维素分子中没有引入新的基团,仍然是典型的植物纤维纤维素红外图谱。随压榨压力的升高,纤维素红外结晶指数增大,分别为 90.89%、93.17%、94.41%和95.84%,这说明压力作用使得分子间的氢键发生了重排,且其强度随压力增大而增强。

图6 不同压榨压力条件下再生植物纤维纤维素的红外谱图
2.2.4 红外谱图的氢键拟合分析
图7显示了不同压榨压力下纤维素红外谱图氢键区域拟合,表5列出了氢键拟合结果和分峰的强度。
从表5可知,不同压力作用后,纤维素分子的氢键仍然以分子间氢键O(6)H…O(3')为主导,但是随压榨压力的升高,纤维素的分子内氢键O(2)H…O(6)和O(3)H…O(5)明显向高频移动,而分子间氢键也向高频移动,不过在压力0.4 MPa作用后却向低频移动,这说明分子间发生了重排。随压榨压力的升高,分子间氢键的强度先增强后减弱,分别为 32.59%、53.31%、64.33%、59.04%,而分子内氢键的强度却随压力作用的增大而减小,在压力达到0.4 MPa之后再次增强。这可能是压力作用的增大一方面使得部分分子内氢键发生断裂,转化成为分子间氢键的连接,另一方面导致晶区结合,分子间更加容易接触,纤维素无定形区的自由羟基得以靠近,形成更多的分子间氢键,这一点也在XRD分析过程中得到证实。当压力升高到0.4 MPa以后,由于外力的增强,分子间氢键发生断裂,在单个纤维素分子链上,葡萄糖内部断裂的分子间氢键重新结合成为分子内氢键O(2)H…O(6)和O(3)H…O(5),也就是分子间氢键发生重排向分子内氢键转化。
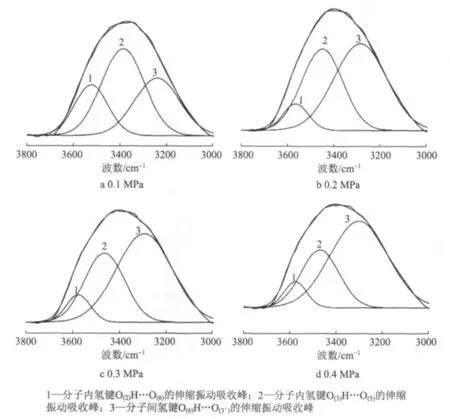
图7 不同压榨压力条件下纤维素红外谱图氢键区域拟合图
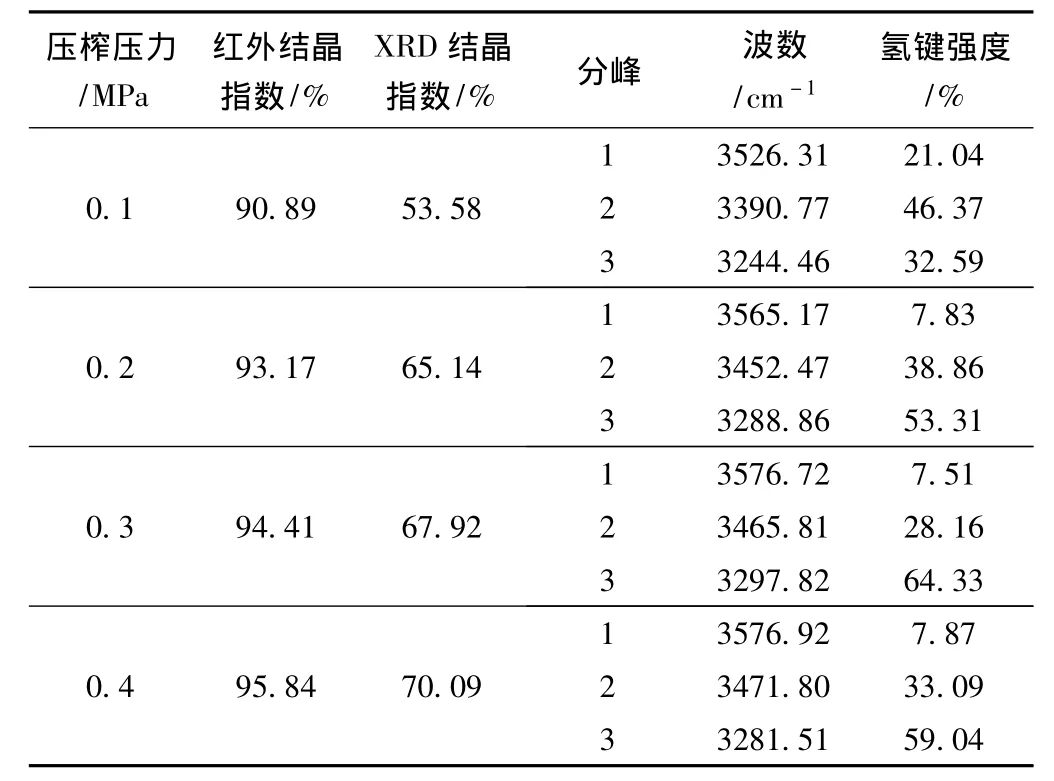
表5 不同压榨条件下红外谱图氢键区域的拟合结果
2.2.5 再生植物纤维纤维素催化水解产糖率分析
图8为不同压榨压力下再生植物纤维纤维素催化水解产糖率的变化。如图8所示,随压榨压力的增加,再生植物纤维纤维素水解率逐渐降低,但葡萄糖产率却呈现先降低后升高的趋势。经过压力作用,纤维素结晶度明显上升,纤维素中分子内氢键向分子间氢键转化,阻碍了催化剂与纤维素的相互作用,从而导致纤维素水解率下降。在经过0.4 MPa处理后,葡萄糖产率略有回升,这可能是纤维素分子中发生了氢键重排,分子内氢键强度增大所致。
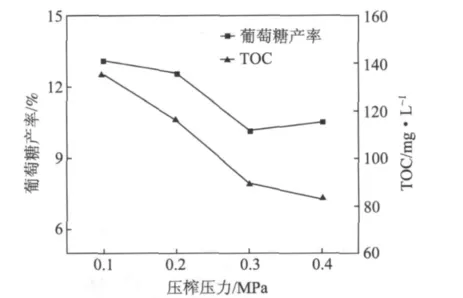
图8 不同压榨压力下再生植物纤维纤维素催化水解产糖率的变化
3 结论
3.1 经不同温度干燥后,纤维素I的基本晶型结构没有改变,也没有新的基团产生或引入,但是结晶度随干燥温度的升高而增大,晶面尺寸从11.60 nm增至17.11 nm,微晶横截面积从 215.18 nm2增至286.76 nm2,结晶区明显增加,这说明温度作用使得纤维素无定形区更加有序,向结晶区转化。
3.2 经过不同压力作用后,纤维素晶体结构没有发生改变,纤维素结晶度由53.58%提高至70.09%,晶面尺寸由11.60 nm增至17.11 nm,微晶横截面积由215.18 nm2增至286.76 nm2,这是压力作用下纤维素晶区间结合所致。
3.3 经过不同干燥温度和压榨压力处理后植物纤维纤维素的水解率均下降,葡萄糖产率也随之降低,只有压力达到0.4 MPa时才略有回升。温度的升高使得纤维素中羟基结合更紧密,排列更有序,催化剂的官能团与纤维素自由羟基不能大量结合;压力增大导致晶区间结合,使得水解过程中纤维素晶体结构的破除难度增大,催化剂的官能团不能有效渗入到分子中,从而造成纤维素水解率下降,影响葡萄糖产率。
3.4 红外谱图氢键区域拟合结果表明,纤维素链间主要靠分子间氢键O(6)H…O(3')结合在一起,稳定纤维素链,而分子内氢键处于辅助地位。不同干燥温度处理后,纤维分子内的结合水被逐渐脱除,表面的自由羟基增加,而自由羟基通过分子内氢键O(2)H…O(6)和O(3)H…O(5)结合,其强度随温度升高而增加;不同压榨压力处理后,分子间氢键的强度先增强后减弱,而分子内氢键变化规律正好相反,主要由于压力作用的增强使得部分氢键断裂重排,分子间氢键向分子内氢键转化。
[1]Nishiyama Yoshiharu,Sugiyama Junji,Chanzy Henri,et al.Crystal Structure and Hydrogen Bonding System in Cellulose Iαfrom Synchrotron X-ray and Neutron Fiber Diffraction[J].J Am Chem Soc,2003,125(47):14300.
[2]Wada Masahisa,Nishiyama Yoshiharu,Langan Paul.X-ray Structure of Ammonia Cellulose I:New Insights into the Conversion of Cellulose I to CelluloseⅢⅠ[J].Macromolecules,2006,39:2947.
[3]Wada Masahisa,Heux Laurent,Nishiyama Yoshiharu,et al.X-ray Crystallographic,Scanning Microprobe X-ray Diffraction,And Cross Polarized/Magic Angle Spinning13C NMR Studies of The Structure of CelluloseⅢⅡ[J].Biomacromolecules,2009,10:302.
[4]Focher B,Palma M T,Canetti M,et al.Structural differences between non-wood plant celluloses:Evidence from solid state NMR,vibrational spectmscopy and X-ray diffractometry[J].Industrial Crops Products,2001,13:193.
[5]Carmen-Mihaela Popescu,Ghita Singurel,Maria-Gristina Popescu,et al.Vibrational spectroscopy and X-ray diffraction methods to establish the differences between hard wood and softwood[J].Carbohydrate Polymers,2009,77:851.
[6]詹怀宇.纤维化学与物理[M].北京:科学出版社,2005:128.
[7]Ye Daiyong,Farriol X.Improving accessibility and reactivity of celluloses of annual plants fort theynthesis of methylcellulose[J].Cellulose,2005,12(5):507.
[8]Marechal Y,Chanzy H.The hydrogen bond network in Iβcellulose as observed by infrared spectrometry[J].Journal of Molecular Structure,2000,523:183.
[9]Sugiyama Junji,Persson Jan,Chanzy Henri.Combined infrared and electron diffraction study of the polymorphism of native celluloses[J].Macromolecules,1991,24:2461.
[10]Kondo T,Sawatari C.A Fourier transform infra-red spectroscopic analysis of the character of hydrogen bonds in amorphous cellulose[J].Polymer,1996,37:393.
[11]Bhuiyan M,Hirai N,Sobue N.Changes of crystallinity in wood cellulose as a mechanism for hornification of bleached kraft pupl[J].Cellulose,2004,11(1):45.
[12]Wistara N,Zhang X J,Young R A.Properties and treatments of pulps from recycled paper.Part II.Surface properties and crystallinity of fibers and fines[J].Cellulose,1999,6(4):325.
[13]Schwanninger M,Rodrigues J C,Pereirac H,et al.Effects of short time vibratory ball milling on the shape of FT-IR spectra of wood and cellulose[J].Vibrational Spectroscopy,2004,36:23.
[14]Struszczyk H.Modification of lignins.III.Reaction of lignosulfonates with chlorophosphazenes[J].Journal of Macromolecular Science A,1986,23:973.
