Genetic diversity and matrilineal structure in Chinese tree shrews inhabiting Kunming, China
2011-12-25CHENShiYiXULingLongBaoYAOYongGang
CHEN Shi-Yi , XU Ling ,3, LÜ Long-Bao , YAO Yong-Gang ,*
(1. Key Laboratory of Animal Models and Human Disease Mechanisms of the Chinese Academy of Sciences & Yunnan Province, Kunming Institute of Zoology, Kunming Yunnan 650223, China; 2. Experimental Animal Core Facility & Kunming Primate Research Center, Kunming Institute of Zoology,the Chinese Academy of Sciences, Kunming Yunnan 650223, China; 3. Graduate School of the Chinese Academy of Sciences, Beijing 100049, China)
Genetic diversity and matrilineal structure in Chinese tree shrews inhabiting Kunming, China
CHEN Shi-Yi1, XU Ling1,3, LÜ Long-Bao2, YAO Yong-Gang1,*
(1.Key Laboratory of Animal Models and Human Disease Mechanisms of the Chinese Academy of Sciences & Yunnan Province,Kunming Institute of Zoology,Kunming Yunnan650223,China; 2.Experimental Animal Core Facility & Kunming Primate Research Center, Kunming Institute of Zoology,the Chinese Academy of Sciences, Kunming Yunnan650223,China; 3.Graduate School of the Chinese Academy of Sciences, Beijing100049,China)
Due to their special phylogenetic position in the Euarchontoglires and close affinity to primates, tree shrews have been proposed as an alternative experimental animal to primates in biomedical research. However, the population genetic structure of tree shrews has largely remained unknown and this has hindered the development of tree shrew breeding and selection. Here we sampled 80 Chinese tree shrews (Tupaia belangeri chinensis) in Kunming, China,and analyzed partial mtDNA control region sequence variation. Based on our samples and two published sequences from northern tree shrews (T. belangeri), we identified 29 substitutions in the mtDNA control region fragment (~ 604 bp)across 82 individuals and defined 13 haplotypes. Seventeen samples were selected for sequencing of the cytochromeb(Cytb; 1134 bp) gene based on control region sequence variation and were analyzed in combination with 34 published sequences to solidify the phylogenetic pattern obtained from control region data. Overall, tree shrews from Kunming have high genetic diversity and present a remarkable long genetic distance to the two reported northern tree shrews outside China.Our results provide some caution when using tree shrews to establish animal models because of this apparent genetic difference. In addition, the high genetic diversity of Chinese tree shrews inhabiting Kunming suggests that systematic genetic investigations should be conducted before establishing an inbred strain for medical and biological research.
Chinese tree shrews; mtDNA; Control region; Cytochormeb; Genetic diversity
Tree shrews (Tupaia belangeri) are small mammals of a squirrel-like appearance formerly placed in the Primates order despite a lack of derived features characteristic of primate species (Sargis, 2004). It is currently placed in the family of Tupaiidae, Scandentia,which together with Dermoptera and Primates, form the group Euarchonta (Murphy et al, 2001; Helgen, 2005).Although the exact phylogenetic position of tree shrews remains resolved, this animal is still regarded as a close relative to primates (Nie et al, 2008; Peng et al, 1991;Seiffert et al, 2003). Given that tree shrews are easy to raise, cheap to maintain, have a high reproductive rate,small body size, and close affinity to primates, this animal may be an alternative to the use of primates in biomedical research. For example, tree shrews have been used in creating animal models for hepatitis B virus and hepatitis C virus infection (Amako et al, 2010; Cao et al,2003; Köck et al, 2001; Ren & Nassal, 2001; Su, 1987;Xu et al, 2007; Yan et al, 1996). Tree shrews were also successfully used in the study of pathogenesis of human myopia and psychosocial stress diseases (Cao et al, 2003;Fuchs, 2005; Norton et al, 2006). In addition, tree shrews have the highest brain to body ratio of all mammals,including humans (Peng et al, 1991; Previc, 2009), which raises the possibility of investigating brain function and creating animal models for human neuro-degenerative diseases.
There are four genera within the family Tupaiidae,consisting ofTupaia,Anathana,Urogale, andDendrogale(Helgen, 2005). In genusTupaia, a total of 15 species are recognized and broadly distributed across Southeast Asia including southern China, India,Philippines, Java, Borneo, Sumatra and Bali (Olson et al,2005; Peng et al, 1991). Chinese tree shrews (Tupaia belangeri) are distributed across southwest China.According to geographical distribution and morphological characteristics, Wang (1987) proposed that Chinese tree shrews could be divided into six subspecies:Tupaia belangeri chinensisdistributed in most of parts of Yunnan, Yun-Gui plateau, and southwest Sichuan;T. b. gaoligongensisdistributed in central and north Gaoligong Mountain;T. b. modestaon Hainan Island;T. b. tonquiniadistributed in southwest Guangxi;T. b. yaoshanensisdistributed in northwest Guangxi; andT. b. yunalisdistributed in central and south Yunnan, northwest Guangxi and southwest Guizhou. Recently, Jia et al (2009) studied the morphometrics of the skull and mandible ofT. b.chinensiscollected in Luquan, Jianchuan, Lijiang, and Yunlong Counties of Yunnan and morphological differences between localities. In combination these studies suggest that Chinese tree shrews show regional differentiation.
Despite growing attention and the strong potential of tree shrews as an animal model for human disease,scant biological data about these species have been collected, especially at the molecular level (Peng et al,1991). This deficit has constrained the utilization of tree shrews as an animal model in biomedical studies.Although the critical problem regarding laboratory breeding and domestication of wild tree shrews was solved more than two decades ago (Peng et al, 1991), an inbred strain of tree shrews has not been established because, among other factors, of a lack of genetic data.
Here we aimed to analyze the genetic structure of tree shrews collected from urban Kunming, Yunnan,China to provide useful information to establish several inbreeding lines from these captured animals. We used mitochondrial DNA (mtDNA) as a genetic marker as it is useful in revealing the origin and demographic history and overall genetic diversity of domestic and wild species (He et al, 2008).
1 Materials and Methods
1.1 Sampling, DNA amplification and sequencing
We collected ear or muscle tissue from 80 Chinese tree shrews (T. b. chinensis) inhabiting the suburb of Kunming, Yunnan, China. Animals were raised at the experimental animal core facility of the Kunming Institute of Zoology and procedures were approved by the Institutional Animal Care and Use Committee of the Kunming Institute of Zoology, the Chinese Academy of Sciences.
Genomic DNA was extracted from tissue samples using the standard phenol/chloroform method. A fragment with a size of 1 488 bp that covers the entire mtDNA control region of Chinese tree shrews was amplified using primer pairs L15356: 5'-CTCAAGGAA GAAGAACAATA-3' / H50: 5'-TGTATGTTTATGGAG TCTATG-3'. Primers were named relative to their
positions in the complete mtDNA sequence of northern tree shrew (GenBank accession number NC_002521).PCR amplification was performed in a volume of 50 μL reaction mixture containing 100 ng of DNA, 10 mmol/L Tris-HCl (pH 8.3), 2.5 mmol/L MgCl2, 50 mmol/L KCl,10 pmol/L of each primer, and one unit of LA Taq polymerase (Takara, Dalian, China) following 35 cycles of 50 s at 94°C, 50 s at 55°C and 60 s at 72°C. The complete mitochondrial Cytbgene was amplified and sequenced in 17 Chinese tree shrews representing different haplotypes as identified by mtDNA control region data. We used primer pair L14058: 5'-ACAAAAACACAATATAAGGCA -3' / H15298: 5'-CTCCGTTTCTGGTTTACAAG -3' to amplify the Cytbgene, applying the same amplification conditions and procedures as used for mtDNA control region. PCR products were purified on spin columns and directly sequenced using Big Dye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Carlsbad, USA) on an ABI 3730 DNA sequencer according to the manufacturer's manual. Two inner sequencing primers(H16093: 5'-CAAGTTAAGTCCAGCTACAAT-3', for mtDNA control region; L15059: 5'-TCCCATTCCTT CACACGTC-3', for Cytbgene) and primers L15356,L14058, and H15298 were used for overlapping sequencing of the analyzed region. The mtDNA sequences of Chinese tree shrew have been deposited in GenBank with accession numbers HQ836245- HQ836324(for mtDNA control region sequences) and HQ836325-HQ836341 (for Cytbgene sequences).
1.2 Data Analysis
We analyzed a fragment of mtDNA control region(~604 bp) in 80 Chinese tree shrews, corresponding to 15416th–16017thregion in mitochondrial genome ofT.belangeri(GenBank accession number NC_002521).Two reported individuals of northern tree shrews (Tupaia belangeri) outside China were retrieved from GenBank and were aligned with those of Chinese tree shrews (Tab.1 and Fig. 1). Sequence variations were exported using Mega 4.0 (Tamura et al, 2007). The available Cytbgene sequences of tree shrews in GenBank, including 32 Chinese tree shrews and two northern tree shrews outside China (Tab. 1), were also analyzed together with 17 newly sequenced samples. Relationships among the lineages were explored using the median-joining networks (Bandelt et al, 1999). Networks were constructed manually and were checked using Network 4.1 (http://www.fluxus-engineering.com/sharenet.htm),in which transitions and transversions were equally weighted. Nucleotide diversity (π) and haplotype diversity (h) were estimated according to Nei (1987) and using DnaSP 5.0 (Librado & Rozas, 2009).
To further determine the phylogenetic position of Chinese tree shrews we retrieved the Cytbgene sequences ofTupaia glis(AY321645),T. longipes(AY321651),T. salatana(AY321654), andDendrogale melanura(AY321634, as the outgroup) from GenBank and constructed a neighbor-joining (NJ) tree using Mega 4.0 (Tamura et al, 2007). The maximum composite likelihood nucleotide substitution model was employed.The rate variation among sites, with four rate categories for the nucleotide gamma shape parameter, wasestimated using jModelTest 0.1.1 (Guindon & Gascuel,2003) which finally yielded the alpha value 0.64.
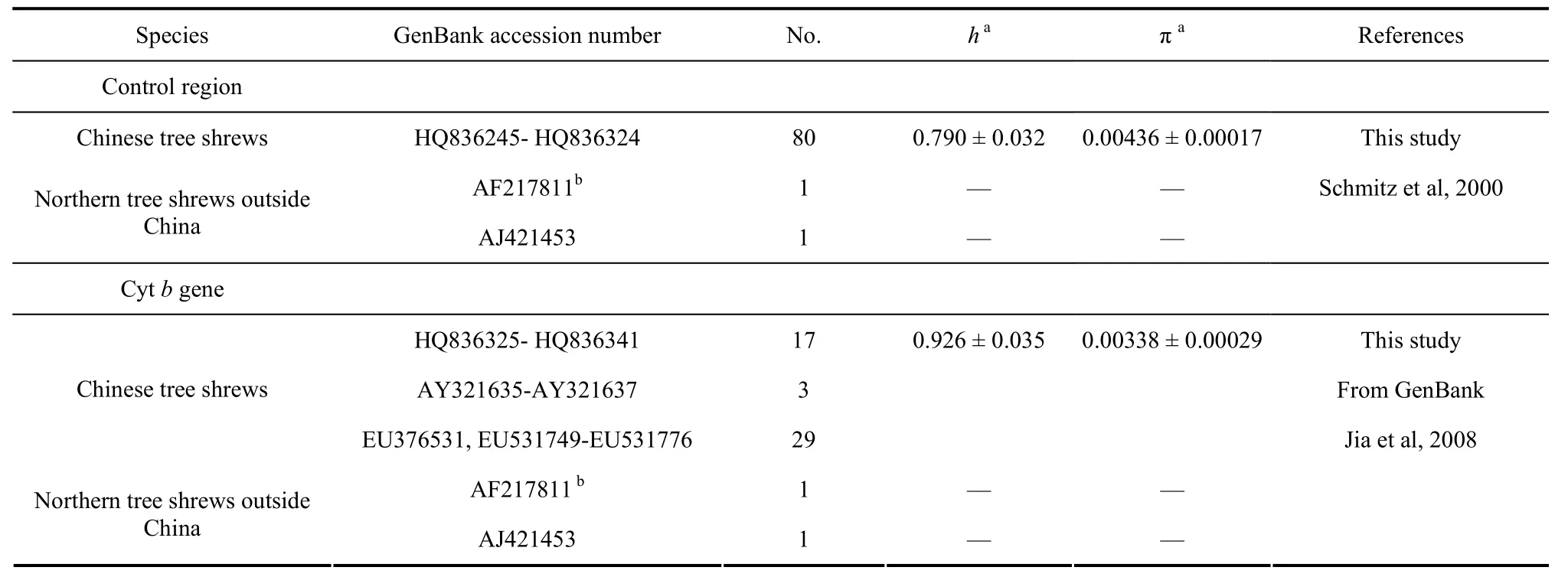
Tab. 1 mtDNA sequence information for tree shrews analyzed in this study
2 Results
2.1 Sequence variation and haplotype distribution
The mtDNA control region sequences of tree shrews presented length mutations, which were caused by insertion/deletion (indels) of tandem repeat or length mutations of a stretch of duplicated nucleotide in region 15449–15456. One sample (TS07) had an extra insertion of CATT repeat in region 15557–15564 which contained two tandem repeats in the analyzed fragment (~604 bp).After the indels were excluded, we found 29 sequence variations in the analyzed fragment of mtDNA control region, which defined 13 haplotypes (H1–H13; Fig. 1).Among them, haplotype H5 was dominant and occurred in 30 individuals, followed by haplotypes H1 (in 14 individuals), H2 (in 12 individuals) and H12 (in 8 individuals). The other haplotypes had a low frequency and were shared by no more than four samples. The two reported northern tree shrews outside China shared haplotype H13, and presented strong differences to the haplotypes identified in Chinese tree shrews (Fig. 1). The overall genetic diversity of 80 Chinese tree shrews (π =0.00436 ± 0.00017 andh =0.790±0.032) was relatively high.
One hundred and twelve variable sites were found in 51 Cytbgene sequences (including 49 Chinese tree shrews and the two northern tree shrews outside China),which defined 26 haplotypes (h1–h26). Among these sequence variations, 17 substitutions caused amino acids changes. Some representative samples bearing different mtDNA control region haplotypes, e.g. H5 and H7, were found to share identical Cytbsequence of h4. However,the two randomly selected individuals sharing mtDNA control region haplotype H5 differed from each other by one substitution in the Cytbsequences (h4 and h5).Samples bearing unique mtDNA control region haplotypes (H1, H2, H3, H8, H9, H12, and H13) also differed from each other by some nucleotide substitutions in the Cytbgene. Remarkably, the two northern tree shrews outside China shared the same Cytbsequence and presented at least 89 mutation distances to Chinese tree shrews (Fig. 2). The nucleotide diversity of 49 Chinese tree shrew Cytbgene sequences was 0.00333 ± 0.00032.
2.2 Network profiles of mtDNA haplotypes in Chinese tree shrews
Fig. 3a shows the network file of mtDNA control region haplotypes in 82 tree shrews analyzed in this study. The network of all Chinese tree shrews followed a roughly star-like pattern, with haplotype H3 residing in the central location and being the nodal type. However,the frequency (5%, 4/80) of H3 was quite low in our samples. All other haplotypes differed from H3 by no more than a 3-mutation distance, suggesting relatively recent divergence. The three dominant haplotypes (H1,H2, and H5) were distributed at the terminal branches of the network. Up to 18-mutation distance existed between Chinese tree shrews (haplotypes H1-H12) and the reported northern tree shrews outside China (haplotype H13).
The network profile of the Cytbhaplotypes followed a similar pattern observed for mtDNA control region sequences (Fig. 3b). Because we selected the samples for Cytbsequencing based on the haplotypes identified by control region sequence information, an estimation of the frequency of each Cytbhaplotype may be biased, despite a fact that we took 32 published Chinese tree shrews samples into consideration.Nonetheless, the phylogenetic relationship as indicated by the network of Cytbhaplotypes might offer further information for us to discern the genetic difference between different lineages. Consistent with the pattern of mtDNA control region sequence data, haplotype h3 was also located in the central location of the network,suggesting its ancestral status. The difference of Cytbgene nucleotide changes between Chinese tree shrews and the reported northern tree shrews (h26) was remarkable, with a ratio (89/1134) even higher than that of the mtDNA control region (18/604).

Fig. 2 Sequence variation of mtDNA cytochrome b gene fragments (1134 bp) of 17 Chinese tree shrews newly sequenced in this study and 34 individuals from GenBank
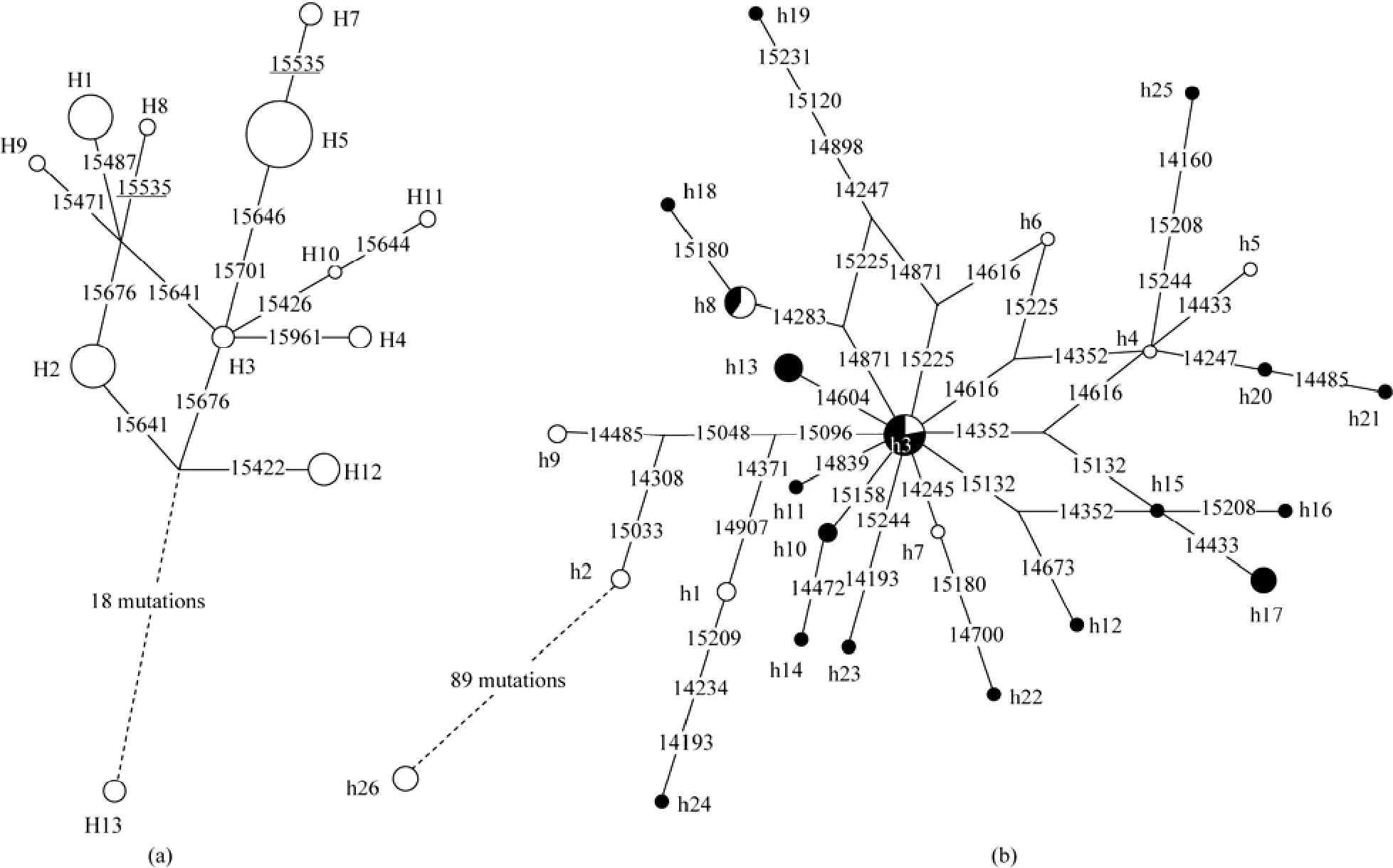
Fig. 3 Network profiles of the mtDNA control region (a) and Cyt b gene (b) haplotypes of tree shrews
2.3 Phylogenetic position of Chinese tree shrews
We constructed an NJ tree of the 26 Cytbhaplotypes of Chinese tree shrews, together with four species from GenBank (Tupaia glis,Tupaia longipes,Tupaia salatana, andDendrogale melanura), to further demonstrate the affinity of Chinese tree shrews to other species (Fig. 4). All Chinese tree shrews haplotypes were clustered together with high bootstrap support. Chinese tree shrews are clearly separated from northern tree shrews outside China and other species, but presented an overall closer relationship to northern tree shrews.

Fig. 4 The neighbor-joining (NJ) tree of the Cyt b gene of 26 Tupaia belangeri haplotypes and four other species (T.glis, T. longipes, T. salatana, and Dendrogale melanura)
3 Discussion
Tree shrews are received more and more attention due to their potential use as animal models for human disease and as alternatives for primates in biomedical research (Cao et al, 2003; Peng et al, 1991). Although the exact phylogenetic position of tree shrews within the Euarchontoglires remains of debate (Arnason et al, 2002;Murphy et al, 2001; Nie et al, 2008; Olson et al, 2005;Peng et al, 1991; Sargis, 2004; Schmitz et al, 2000),available evidence has undoubtedly suggested a closer affinity of tree shrews to well-known primates such as rhesus monkeys (Macaca mulatta) and crab-eating macaques (Macaca fascicularis) than mice or rats. Given that tree shrews are also small animals and easy to raise,it may be promising to develop them as replacements for monkeys and other endangered non-human primate species.
There are 15 recognized species and 40 subspecies in genusTupaia(http://www.bucknell.edu/msw3/; Helgen,2005). However, to our knowledge an insufficient number of molecular studies have been done and necessary information for biomedical research and breeding as an experimental animal remain unknown.Work on gene expression in the hippocampal tissue of tree shrews treated with cortisol (Alfonso et al, 2004)and proteome analysis of aflatoxin B1-induced hepatocarcinogenesis (Li et al, 2008) has yielded useful information, but compared to the available knowledge for mice and rats there is still much unknown about these animals.
Here we focused on mtDNA sequence variation in Chinese tree shrews captured from urban Kunming. Our initial aim was to better understand the genetic structure of this species and to establish a matrilineal marker for tracing lineages in a subsequent tree shrews breeding project. In addition, we wanted to better understand overall genetic diversity of this animal resource. By comparing the mtDNA sequence differences between Chinese tree shrews and northern tree shrews, we found that they had a large divergence (Figs.1, 2). Network analyses showed that Chinese tree shrews and northern tree shrews are clearly separated from each other (Fig. 3).Our results raise caution over the routine use of tree shrews as animal models and genetic differences are ignored, especially when studies simply named their subjects asTupaia belangeri.
The divergence of Chinese tree shrews and northern tree shrews was unexpected and implies that a systematic analysis of the six proposed subspecies of Chinese tree shrew (Wang, 1987) requires verification in future studies. Moreover, capturing and domesticating wild animals with the representative lineages should be encouraged during our efforts in establishing inbred tree shrew strains for medical and biological research. The observation that the tree shrew population inhabiting Kunming has a considerably high mtDNA genetic diversity is consistent with the result of a recent study of microsatellite markers in which He et al (2009) found that nine out of 11 loci exhibited polymorphisms in 30 Chinese tree shrews. Data such as that presented here will be helpful in selecting founding individuals for the establishment of an inbred strain of Chinese tree shrews.
Acknowledgement:We thank YANG Zhao-Qing for collecting some tree shrews samples and JIANG Xue-Long for comments on an early version of the manuscript.
Alfonso J, Aguero F, Sanchez DO, Flugge G, Fuchs E, Frasch AC,Pollevick GD. 2004. Gene expression analysis in the hippocampal formation of tree shrews chronically treated with cortisol [J].J Neurosci Res, 78(5): 702-710.
Amako Y, Tsukiyama-Kohara K, Katsume A, Hirata Y, Sekiguchi S,Tobita Y, Hayashi Y, Hishima T, Funata N, Yonekawa H, Kohara M. 2010. Pathogenesis of hepatitis C virus infection inTupaia belangeri[J].J Virol, 84(1): 303-311.
Arnason U, Adegoke JA, Bodin K, Born EW, Esa YB, Gullberg A,Nilsson M, Short RV, Xu XF, Janke A. 2002. Mammalian mitogenomic relationships and the root of the eutherian tree [J].Proc Natl Acad Sci U S A, 99(12): 8151-8156.
Bandelt H-J, Forster P, Röhl A. 1999. Median-joining networks for inferring intraspecific phylogenies [J].Mol Biol Evol, 16(1): 37-48.
Cao J, Yang EB, Su JJ, Li Y, Chow P. 2003. The tree shrews: adjuncts and alternatives to primates as models for biomedical research [J].J Med Primatol, 32(3): 123-130.
Fuchs E. 2005. Social stress in tree shrews as an animal model of depression: an example of a behavioral model of a CNS disorder[J].CNS Spectr, 10(3): 182-190.
Guindon S, Gascuel O. 2003. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood [J].Syst Biol,52(5): 696-704.
He BL, Shen PQ, Chen LL, Jia JL, Liu RW, Li B, Zheng H, Li ML.2009. Polymorphism microsatellites in tree shrews (Tupaia Belangeri Chinensis) [J].Acta Lab Anim Sci Sin, 17(2): 143-145.(in Chinese)
He DQ, Zhu Q, Chen SY, Wang HY, Liu YP, Yao YG. 2008. A homogenous nature of native Chinese duck matrilineal pool [J].BMC Evol Biol, 8: 298.
Helgen KM. 2005. Order Scandentia[M]// Wilson DE, Reeder DM (ed).2005. Mammal Species of the World: A Taxonomic and Geographic Reference. 3rd ed. Maryland: Johns Hopkins University Press, 104-109.
Jia T, Lin AQ, Wang R, Zhu WL, Xiao CH, Liu CY, Meng LH, Lian X,Wang ZK. 2009. Pilot study ofTupaia belangerifrom Yunnan Province based on morphometrics of the skulls and mandibles [J].Acta Theriol Sin, 29(3): 259-268. (in Chinese)
Jia T, Yang XM, Li ZH, Zhu WL, Xiao CH, Liu CY, Wang ZK. 2008.Classified significance ofTupaia belangerifrom Luquan District,Kunming based on Cytbgene sequences [J].Chn J Zool, 43(4):26-33. (in Chinese)
Köck J, Nassal M, MacNelly S, Baumert TF, Blum HE, von Weizsäcker F. 2001. Efficient infection of primary tupaia hepatocytes with purified human and woolly monkey hepatitis B virus [J].J Virol, 75(11): 5084-5089.
Li Y, Qin X, Cui J, Dai Z, Kang X, Yue H, Zhang Y, Su J, Cao J, Ou C,Yang C, Duan X, Yue H, Liu Y. 2008. Proteome analysis of aflatoxin B1-induced hepatocarcinogenesis in tree shrew (Tupaia belangeri chinensis) and functional identification of candidate protein peroxiredoxin II [J].Proteomics, 8(7): 1490-1501.
Librado P, Rozas J. 2009. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data [J].Bioinformatics, 25(11):1451-1452.
Murphy WJ, Eizirik E, O'Brien SJ, Madsen O, Scally M, Douady CJ,Teeling E, Ryder OA, Stanhope MJ, de Jong WW, Springer MS.2001. Resolution of the early placental mammal radiation using Bayesian phylogenetics [J].Science, 294(5550): 2348-2351.
Nei M. 1987. Molecular Evolutionary Genetics [M]. New York:Columbia University Press.
Nie W, Fu B, O'Brien PC, Wang J, Su W, Tanomtong A, Volobouev V,Ferguson-Smith MA, Yang F. 2008. Flying lemurs—the 'flying tree shrews'? Molecular cytogenetic evidence for a Scandentia-Dermoptera sister clade [J].BMC Biol, 6: 18.
Norton TT, Amedo AO, Siegwart JT, Jr. 2006. Darkness causes myopia in visually experienced tree shrews [J].Invest Ophthalmol Vis Sci,47(11): 4700-4707.
Olson LE, Sargis EJ, Martin RD. 2005. Intraordinal phylogenetics of treeshrews (Mammalia : Scandentia) based on evidence from the mitochondrial 12S rRNA gene [J].Mol Phylogenet Evol, 35(3):656-673.
Peng YZ, Ye ZZ, Zou RJ, Wang YX, Tian BP, Ma YY, Shi LM. 1991.Biology of Chinese tree shrews (Tupaia belangeri chinensis) [M].Kunming: Yunnan Science and Technology Press. (in Chinese)
Previc FH. 2009. The Dopaminergic mind in human evolution and history [M]. Cambridge, New York, Melbourne, Madrid, Cape Town, Singapore, São Paulo, Delhi: Cambridge University Press.
Ren S, Nassal M. 2001. Hepatitis B virus (HBV) virion and covalently closed circular DNA formation in primary tupaia hepatocytes and human hepatoma cell lines upon HBV genome transduction with replication-defective adenovirus vectors [J].J Virol, 75(3):1104-1116.
Sargis EJ. 2004. New views on tree shrews: the role of tupaiids in primate supraordinal relationships [J].Evol Anthropol, 13(2): 56-66.
Schmitz J, Ohme M, Zischler H. 2000. The complete mitochondrial genome ofTupaia belangeriand the phylogenetic affiliation of Scandentia to other eutherian orders [J].Mol Biol Evol, 17(9):1334-1343.
Seiffert ER, Simons EL, Attia Y. 2003. Fossil evidence for an ancient divergence of lorises and galagos [J].Nature, 422(6930): 421-424.
Su JJ. 1987. Experimental infection of human hepatitis B virus (HBV)in adult tree shrews [J].Chn J Pathol, 16(2): 103-106, 122. (in Chinese)
Tamura K, Dudley J, Nei M, Kumar S. 2007. MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0 [J].Mol Biol Evol, 24(8): 1596-1599.
Wang YX. 1987. Taxonomic research on Burma-Chinese tree shrew,Tupaia belangeri(Wagner), from Southern China [J].Chn J Zool,8(3): 213-230. (in Chinese)
Xu X, Chen H, Cao X, Ben K. 2007. Efficient infection of tree shrew(Tupaia belangeri) with hepatitis C virus grown in cell culture or from patient plasma [J].J Gen Virol, 88: 2504-2512.
Yan RQ, Su JJ, Huang DR, Gan YC, Yang C, Huang GH. 1996.Human hepatitis B virus and hepatocellular carcinoma. I.Experimental infection of tree shrews with hepatitis B virus [J].J Cancer Res Clin Oncol, 122: 283-288.
昆明城郊中国树鼩群体线粒体DNA遗传多样性
陈仕毅1, 许 凌1,3, 吕龙宝2, 姚永刚1,*
(1. 中国科学院和云南省动物模型与人类疾病机理重点实验室, 中国科学院昆明动物研究所, 云南 昆明 650223;2. 中国科学院昆明动物研究所 实验动物中心, 云南 昆明 650223; 3. 中国科学院研究生院, 北京 100049)
由于树鼩是灵长类动物的近亲, 且具有体型小、繁殖周期短、饲养管理成本低等优点, 长期以来被认为有望替代灵长类动物用于人类疾病的动物模型研究。然而, 目前对树鼩的群体遗传结构还知之甚少, 这极大地限制了其在疾病动物模型研究的应用, 也是其品系资源创制的瓶颈。本研究通过分析80只采自于云南省昆明周边地区的野生树鼩 (Tupaia belangeri chinensis) 线粒体DNA (mtDNA) 多态性, 结合国外报道的2个树鼩 (Tupaia belangeri)
序列比较后发现, 在604 bp的mtDNA控制区片段中共检测到29个核苷酸替代变异, 这些变异共界定了13种单倍型, 表现较高的群体遗传多样度。另外, 昆明地区的树鼩与国外报道的2个树鼩间存在较大的遗传分化, mtDNA控制区单倍型之间的核苷酸替换数大于18个, 远高于昆明地区树鼩群体内部不同单倍型之间的差异。选择含有代表性的mtDNA控制区单倍型的17个昆明地区树鼩个体进一步测定了细胞色素b基因片段 (1134 bp), 结合前人报道的数据分析, 结果进一步支持 mtDNA控制区数据反映的遗传格局及揭示的昆明地区树鼩与国外报道树鼩之间的明显差异。本研究结果提示, 昆明地区树鼩与国外树鼩之间存在较大遗传差异, 在将树鼩用于人类疾病动物模型研究中要注意这些遗传差别。昆明城郊的树鼩群体具有较高的遗传多样度, 在开展近交系建立等工作时须考虑选取群体内部具有代表性的mtDNA世系。
2010-12-02;接受日期:2010-12-27
中国树鼩; 线粒体DNA; 高变区; 细胞色素b; 遗传多样性
Q959.832; Q951.3; Q347
A
0254-5853-(2011)01-0017-07
10.3724/SP.J.1141.2011.01017
date: 2010-12-02; Accepted date: 2010-12-27
s: This study was supported by grants from Yunnan Province (2009CI119), the Chinese Academy of Sciences (KSCX2-EW-R-11 and KSCX2-EW-J-23) and the Key Laboratory of Animal Models and Human Disease Mechanisms of the Chinese Academy of Sciences& Yunnan Province*
(通信作者), Email: ygyaozh@gmail.com
