碳酸盐岩系有机氮与无机氧热化学交换作用研究①
2011-12-15丁康乐虞启明
丁康乐 王 辉 罗 跃 杨 欢 虞启明
(1.长江大学化学与环境工程学院 湖北荆州 434023;2.大庆油田勘探开发研究院 黑龙江大庆 163712; 3.格里菲斯大学工程学院 昆士兰 澳大利亚 4111)
碳酸盐岩系有机氮与无机氧热化学交换作用研究①
丁康乐1王 辉2罗 跃1杨 欢1虞启明3
(1.长江大学化学与环境工程学院 湖北荆州 434023;2.大庆油田勘探开发研究院 黑龙江大庆 163712; 3.格里菲斯大学工程学院 昆士兰 澳大利亚 4111)
深埋碳酸盐岩储层中的水与金属氧化物可能会影响到吡咯类含氮化合物的保存。对吡咯-水-氧化铝反应体系进行了热模拟实验研究,根据模拟实验结果,探讨了反应机理,并考察了反应的动力学特征。结果表明,吡咯-水-氧化铝体系可以发生反应,产物主要为呋喃与氨气,升高温度对反应有利。氧化铝对有机氮与无机氧热化学交换作用起到了明显的催化作用,含水量增大不利于反应的进行。在氧化铝存在条件下,吡咯转化为呋喃的反应活化能为109.35 kJ/mol。
碳酸盐岩储层 吡咯-水-氧化铝反应体系 热模拟实验 动力学 反应机理
在我国,碳酸盐岩分布广泛,沉积厚度大,具有良好的油气勘探前景[1,2]。随着勘探程度的提高,石油天然气勘探正逐渐走向深部[3]。深埋石油地质体中的吡咯类化合物,蕴藏着丰富的地质-地球化学信息,是当今国际地学的研究热点[4~10]。关于油气储层中吡咯类化合物的研究文献主要发表在近20年当中。在吡咯类化合物的分离和鉴定、吡咯类化合物在油气运移方面的研究和应用、吡咯类化合物的组成和分布[4~14]以及吡咯类化合物的成因[15~19]等4个主要方向上,国内外学者进行了大量研究,并且取得了一些有意义的认识,但仍有许多理论和实践方面的关键问题需要进一步探讨,特别是对于深部储层中吡咯类化合物保存与演化的研究目前还处于起步阶段。
沉积盆地是一个巨大的地温热化学反应器,近年来,有机地球化学家开始认识到地下化学环境对油气形成和组成有着非常重要的影响[20,21]。油气地球化学家已经发现,碳酸盐岩储集层中的水[22~26]、金属氧化物[20,21]、过渡金属[27~29]、无机盐类[30,31]以及黏土矿物[32~34]等无机因素可影响到油气的生成和演化。理论上,吡咯类化合物与烃类一样受控于上述多种地质-地球化学因素,但在以往研究吡咯类化合物的过程中,对沉积环境中一些可能影响到吡咯类化合物保存的无机因素,特别是关于吡咯类含氮化合物与水之间的有机-无机相互作用,国际地学领域还未见有这方面的报导。针对我国海相碳酸盐岩普遍存在演化程度高、有机质丰度低、分子地球化学信息少等特点,这一研究领域内所取得的新认识,将在油气运移、油源对比、碳酸盐岩油气成熟度以及深埋碳酸盐岩沉积环境特征探讨等方面有广泛的应用前景,对寻找在高过成熟条件下依然有效的新指标以及研究深层地质体中的氮循环具有重要的理论和实际价值。
本文以吡咯作为吡咯类含氮化合物的模型化合物,首次对吡咯-水-氧化铝反应体系进行了热模拟实验研究。通过色谱-质谱(GC-MS)和傅立叶变换红外(FT-IR)等分析技术对实验结果进行了分析,提出了有机氮与无机氧热化学交换作用发生的途径,在此基础上考察了反应过程的动力学特征,求出了动力学参数,并对反应机理进行了初步的探讨。
1 实验部分
1.1 实验装置与实验条件
实验装置主要由200 ml高压反应釜、气路和取样分析系统组成。反应釜为江苏海安石油科研仪器有限公司WYF-1型高压釜,控温精度为±1℃。每次实验时,将0.1 g氧化铝粉末置于釜底,密闭高压釜并抽真空后,从进料管处向釜内加入10.0 ml吡咯与10.0 ml蒸馏水。吡咯为分析纯,由美国Sigma-Aldrich公司提供,纯度为99%。氧化铝与酚酞试纸由北京益利精细化学品公司以及天津大茂化学试剂厂提供。气相色谱分析用的呋喃标样为分析纯,纯度为99%,由香港Farco chemical supplies公司提供。高纯NH3(99.999%)标气由武汉赛尔气体有限公司提供。
吡咯-水-氧化铝反应体系热模拟反应温度为350℃,375℃,400℃,425℃和450℃,反应体系的最终压力因温度而异,一般在6.0~10.0 MPa范围内。由于低温时反应较难进行,室温到300℃时对反应釜采取满负荷直接加热的方法。根据动力学计算模型参数β的要求,300℃到最终的反应温度采取程序升温的方法:300~350℃,360 h;300~375℃,288 h; 300~400℃,216 h;300~425℃,144 h;300~450℃,72 h。待达到设定时间后,将高压釜迅速从加热炉中取出,在空气中冷却30 min,再用自来水喷淋釜体使其迅速冷却至室温。打开釜盖,用移液管抽出釜中有机相与水相混合液,再用蒸馏水冲洗釜壁3~4次,每次蒸馏水用量10 m l。用微型分液漏斗对有机相与水相混合液进行液-液分离。有机相产物用傅立叶变换红外光谱仪与气相色谱-质谱联用仪分析其组成。水相以酸度计检测反应体系pH变化,并用美国Agilent 6890气相色谱仪对其挥发组分进行分析。
1.2 分析方法
反应后有机相产物组成采用美国 Thermo-Finnigan Trace-DSQ气相色谱-质谱联用仪分析。色谱条件:汽化室温度260℃,HP-5MS弹性石英毛细柱(30 m X0.25 mm X 0.25μm),载气为高纯He气(99.99%),载气流速1 mL/min。进样口温度290℃,柱温300℃。柱升温程序:初温40℃,恒温2 min,以4℃/min升温至200℃,保持2 min,再以2.5℃/min升温至300℃并保持15 min。质谱条件:电子轰击离子源(EI);电力电压70 eV;离子源温度:200℃;灯丝电流100μA,电子倍增器电压1 200 V;全扫描质量范围35~500 amu。实验数据处理由Xcalibur软件系统完成,未知化合物经计算机检索同时与NIST 05谱库和Wiley 6.0谱库相匹配,仅当匹配度和纯度大于800(最大值1000)的鉴定结果才予以报道。按峰面积归一化法计算各组分的相对含量。
反应后水相产物的挥发组分利用美国Agilent 6890气相色谱仪进行分析。Tekmar 3100型吹扫捕集浓缩器,火焰离子化检测器(FID)与色谱工作站,均为美国惠普公司生产。HP-624色谱柱(30 m X 0.53 mm X 3.0μm);柱温度250℃,进样口温度200℃;升温程序35~150℃(10℃/min)保持4 min,100~250℃(5℃/min)保持4 min。分流进样,分流比30∶1,高纯氮载气,载气流量7 ml/min。尾吹气流速25 ml/min。柱前压:60 kPa。气密型注射器(澳大利亚SGE公司)。顶空瓶和内衬聚四氟乙烯膜的硅橡胶垫由美国Supelco公司提供。
FT-IR测试采用美国Nicolet公司MAGNAIR560E.S.P型傅立叶变换红外光谱仪,波数范围4 000~400 cm-1,扫描次数32,分辨率4 cm-1。
上海康仪智能型酸度计PHS-3C,pH测量范围为0.00~14.00,测量误差控制在±0.01。
2 结果与讨论
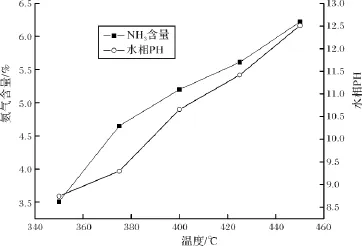
图1 氨气含量与水相pH随温度变化曲线Fig.1 The content of ammonia and the pH ofwater phase with different temperatures
2.1 产物分析
当反应结束后,以润湿的酚酞试纸对反应体系中水相的酸碱度进行定性分析,发现酚酞试纸变粉红色,由于实验所用的反应物吡咯以及产物呋喃均不能使润湿的酚酞试纸发生颜色改变[36],这表明反应前后体系水相酸碱度已发生明显改变,且呈强碱性。进一步利用Agilent 6890气相色谱仪对反应后水相产物中的挥发组份进行了分析。设定气相色谱仪,使之处于稳定运行状态。用注射器取约8 ml水相产物,置于25 ml顶空瓶中,并迅速盖紧瓶盖后置于70℃的油浴槽中平衡1 h。通过气相色谱的顶空进样器抽取顶空瓶内液面上部气体50μl,将样品注入气相色谱仪进行测定。发现水相产物中明显有氨气生成,氨气含量以及水相pH随温度变化关系,结果见图1。由图1可知,氨气含量与水相pH均随温度升高而增大,说明高温促进了氨气的生成。氨气生成的主要途径是可能是吡咯分子中的有机氮原子被水中的无机氧原子置换后形成的。
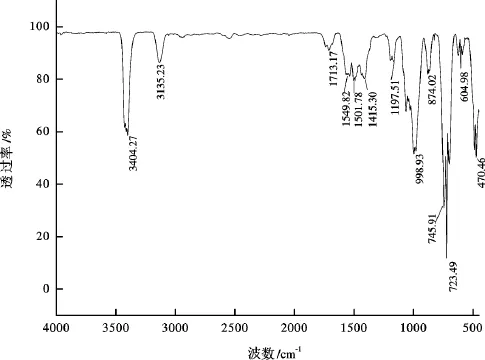
图2 吡咯-水-氧化铝反应体系中有机相产物的FT-IR谱图(450℃)Fig.2 FT-IR spectrum of organic products in the pyrrole-H2 O-Al2 O3 system
通过傅立叶变换红外光谱仪(FT-IR)对反应后的有机相产物进行了初步定性表征。450℃时吡咯-水-氧化铝反应体系中有机相产物的FT-IR谱图见图2,吡咯和呋喃的标准谱图见图3和图4。在呋喃的标准谱图中,3 141.64 cm-1峰是呋喃环C-H键的伸缩振动,1 578.65 cm-1峰、1 479.36 cm-1峰和1 373.67 cm-1峰为呋喃环骨架振动。1 191.10 cm-1峰是C-H键变角振动,1 066.19 cm-1峰是C-O键伸缩振动。1005.34 cm-1峰是呋喃环非对称伸缩振动。874.02 cm-1峰是呋喃环C-H键面外弯曲振动,749.11 cm-1峰是呋喃环弯曲振动,604.98 cm-1峰是呋喃环变形振动,是呋喃的特征频率[35]。在吡咯的标准谱图中,3401 cm-1峰是N-H键伸缩振动, 3 132.03 cm-1峰是吡咯环 C-H键伸缩振动, 1 537.01 cm-1峰是吡咯环对称伸缩振动,由吡咯环上===C C伸缩振动引起,1415.30 cm-1峰是吡咯环的不对称伸缩振动,由吡咯环上C-C键伸缩振动引起,995.73 cm-1峰是C-N键的伸缩振动,720.29 cm-1峰是吡咯环弯曲振动,473.66 cm-1峰是N-H键面外弯曲振动[35]。上述的特征频率在反应后有机相产物的FT-IR谱图(图2)中都有所体现。可见有机相中有呋喃生成。
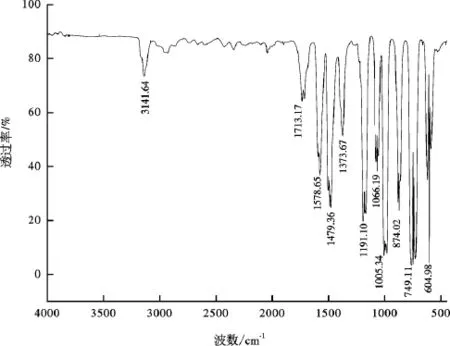
图4 呋喃的标准FT-IR谱图Fig.4 Standard FT-IR spectrum of furan
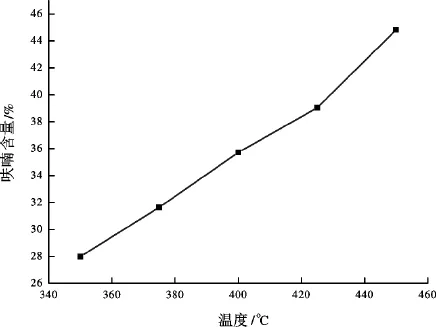
图5 呋喃含量随温度变化关系曲线Fig.5 Change of the reaction conversions under different temperatures
利用气相色谱-质谱联用仪对有机相产物进行了定量分析。根据气相色谱-质谱定量分析结果,得到350~450℃时吡咯-水-氧化铝反应体系有机相产物中呋喃含量分别为27.99%,31.65%,35.72%, 39.04%,44.83%。图5是呋喃含量随温度变化关系曲线。由图5可知,随着温度升高,呋喃含量增加,即吡咯转化率增大,可见温度是影响吡咯转化为呋喃的重要因素,这与水相分析结果一致。
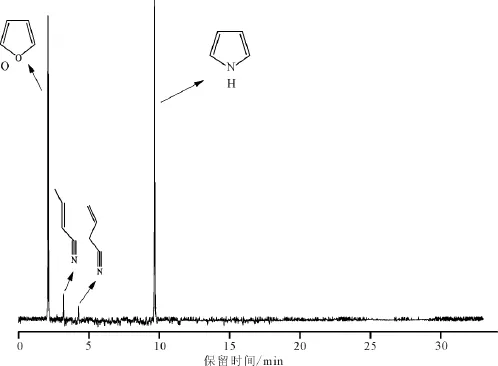
图6 吡咯-水-氧化铝反应体系中有机相产物的总离子流图Fig.6 Total ion chromatogram for organic products in the pyrrole-H2 O-Al2 O3 system(450℃)
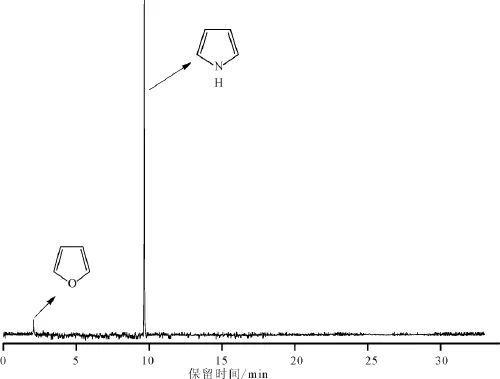
图7 吡咯-水反应体系中有机相产物的总离子流图Fig.7 Total ion chromatogram for organic products in the pyrrole-H2O system(450℃)
图6为450℃时吡咯-水-氧化铝反应体系中有机相产物的总离子流图。根据图6可知,在反应后的有机相组成中,除了吡咯与呋喃外,还有少量的2-丁烯腈与3-丁烯腈生成,它们应该是吡咯在高温和氧化铝作用下形成的开环裂解产物。由于2-丁烯腈与3-丁烯腈的含量均低于5%,因此反应体系中主要发生的是吡咯与水之间的有机氮与无机氧热化学交换作用。由反应后水相及有机相产物分析结果可知,模拟试验温度高于350℃时,吡咯-水-氧化铝反应体系发生了明显的化学反应,主要生成了呋喃与氨气。
2.2 反应机理初探
吡咯热分解温度高达650℃以上[37],因此吡咯的环状分子结构具有较高的热稳定性。吡咯-水-氧化铝体系在350℃就可以发生明显的有机氮与无机氧热化学交换作用,反应转化率为27.99%。图7为450℃时吡咯-水反应体系(空白试验)中有机相产物的总离子流图。由空白试验结果可知,反应后有机相产物主要是吡咯与呋喃,反应转化率仅为7.74%,远低于相同温度下吡咯-水-氧化铝体系的反应转化率(44.83%)。这表明模拟实验过程中氧化铝对吡咯生成呋喃的热化学反应过程起到了明显的催化作用。
吡咯生成呋喃的热化学反应过程可能分为以下7个基元步骤:
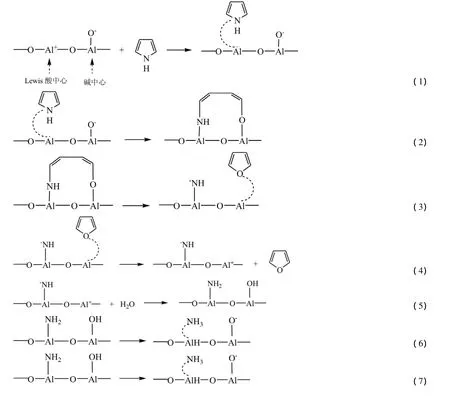
步骤(1)为吡咯吸附过程。吡咯通过其分子中的氮原子占据固体氧化铝表面的Lewis酸中心以达到化学吸附。吡咯分子中的氮原子可能通过单点或多点吸附在氧化铝表面Lewis酸中心上以填补Al上的氧空穴;在步骤(2)中,吡咯分子开环。化学吸附态吡咯中的一个C-N键断裂,形成新的C-O键,从而形成半圆环的吸附结构;步骤(3)为环化过程。半圆环中的C-N键断裂,形成新的C-O键,从而形成呋喃的吸附结构;步骤(4)是呋喃脱附过程。吸附态的呋喃脱附生成呋喃;在步骤(5)中,氧化铝固体表面吸水。氧化铝表面上不饱和离子NH-与Al+通过吸水反应,形成的-OH和-NH2;步骤(6)形成NH3。H+质子从氧化铝催化剂上的-OH转移到-NH2形成NH3;步骤(7)是NH3解离吸附。氧化铝催化剂的表面的Lewis酸中心恢复,为下一步反应做好准备。由以上步骤(1)~(7)可知,在反应结束后,氧化铝分子结构并未发生改变,说明氧化铝并非是反应物,而只是对整个过程催化作用,其中氧化铝固体表面Lewis酸中心很可能是催化活性位。通过氧化铝,水分子中的氧原子与吡咯分子中的氮原子发生了热化学交换。此外,氨气分子中的两个氢原子均来源于水分子。因此,水实际作为反应物,在氧化铝的催化作用下,与吡咯发生了有机氮与无机氧热化学交换作用。
需要指出的是,上述7个基元步骤的建立,主要是依据反应物与检测到的产物分子结构做出的初步推测。未来对咔唑、苯并咔唑、二苯并咔唑等吡咯类含氮化合物模拟实验工作中,将对上述反应机理进行进一步验证和补充。
2.3 动力学研究
将吡咯生成呋喃的热化学反应视为一级反应,则反应动力学方程可写为[38],
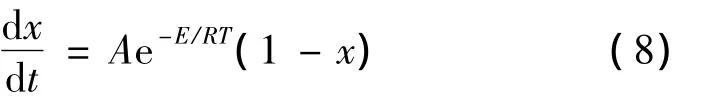
将上式进行积分并整理得到,
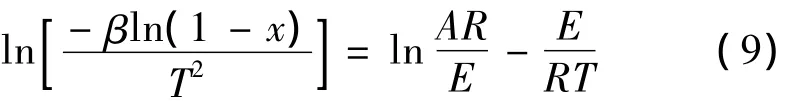
在模拟实验过程中,根据温度与时间可以相互补偿的原则,实验温度要高于实际地质条件下的温度区间。当模拟温度低于200℃时,水相产物酸碱度未发生变化,有机相中也没有检测到呋喃的生成,可能是吡咯环状分子结构具有较高的化学稳定性(吡咯热分解温度高达650℃以上)导致该反应具有较高的活化能。模拟试验温度较低时,在有限的实验时间范围内还不足以使吡咯与水产生明显的化学反应,反应速度很慢,因此在短时间内检测不到相应的反应产物。当实验温度达到350℃时,水相pH发生明显改变,呈较强碱性,色谱检测有氨气产生,有机相伴有呋喃的生成。
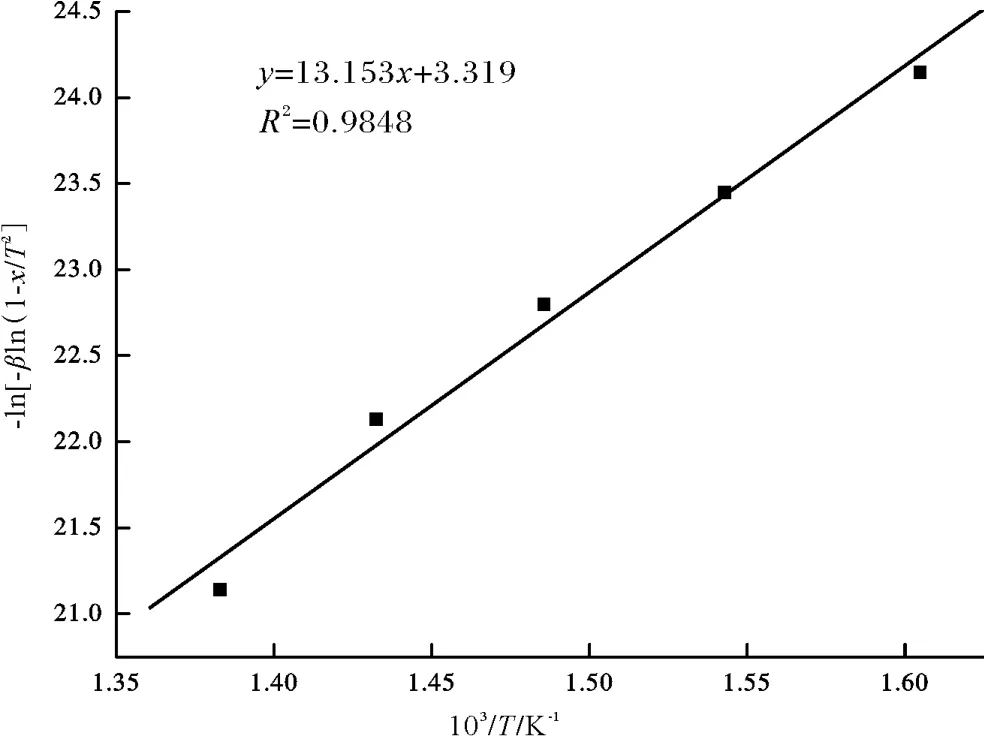
图8 吡咯-水-氧化铝反应的回归直线Fig.8 The regression line for the reaction between pyrrole,water and alumina
根据以往烃源岩的生烃模拟实验,在与本实验相似的高压釜体系中,350~450℃的温度条件下,烃源岩的有机质成熟度Ro值可达到1.6~2.3[39],与此相对应的地质温度大约为110~140℃,这证明了深部碳酸盐岩储层中吡咯-水-氧化铝反应体系可以发生有机氮与无机氧热化学交换作用。
氧化铝与水在高温下存在如下平衡[40]:
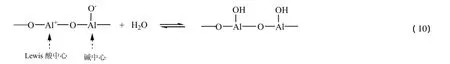
模拟实验体系中含水量仅为10 m l,在高温下可形成不饱和蒸汽,因此部分氧化铝表面会裸露出Lewis酸中心,从而对吡咯转化为呋喃起到催化作用。空白试验表明,相同温度下,随着体系含水量增大,反应转化率逐渐降低,因此深部储层中吡咯类含氮化合物的保存与蚀变过程中,金属氧化物以及含水量是两个重要因素。可以推断,当储层水含量过多时将会钝化金属氧化物的催化活性位,进而抑制吡咯类含氮化合物与水间的有机-无机相互作用程度。根据吡咯-水-氧化铝体系热模拟实验研究结果及反应机理探讨可知,在一定的温度和压力下,深部储层中广泛存在的水在金属氧化物催化作用下,很可能会与吡咯类含氮化合物发生一定程度的化学反应。在此过程中,吡咯类含氮化合物分子中的有机氮被水分子中的无机氧所交换,导致部分吡咯类含氮化合物会部分转化为具有相似骨架结构的含氧非烃化合物,进而影响吡咯类含氮化合物在储层中的组成与分布。
2.4 存在的问题及展望
咔唑、苯并咔唑、二苯并咔唑等吡咯类含氮化合物的分子结构具有较高的热稳定性(一般分解温度>650℃)。前期大量探索性实验表明,它们与水发生有机-无机相互作用所需的实验温度较高(> 450℃),同时反应转化率较低,鉴于目前尚不明确深埋地质环境中催化/抑制等关键因素的前提下,为进行动力学研究,而靠单纯地升高模拟实验温度来获得高反应转化率,极易引起较大规模的热裂解副反应,将不利于考察吡咯类含氮化合物与无机流体间的化学作用机理与动力学特征,因此本文初步选择最简单的吡咯作为吡咯类含氮化合物的模型化合物,氧化铝作为金属氧化物的模型化合物,对吡咯-水-氧化铝反应体系进行了热模拟实验研究。
利用气相色谱-质谱联用仪对有机相产物进行了分析过程中,发现除了吡咯与呋喃外,还有少量的2-丁烯腈与3-丁烯腈,它们应该是吡咯在高温和氧化铝作用下形成的开环裂解产物。由于2-丁烯腈与3-丁烯腈的含量均很低,同时红外光谱准确度大约在1%左右,其灵敏度远低于气相色谱-质谱,因此微量的2-丁烯腈与3-丁烯腈在红外光谱图中未能得到明显的反映。在本文中,红外光谱法可以作为确定产物组成的一个辅助分析方法。
应当指出,与地下储层中的真实条件相比,本文模拟实验中客观地考虑了水、氧化铝等沉积环境中可能影响到吡咯类化合物保存的无机因素,但暂且忽略了膏岩、白云石、黄铁矿等其它矿物质可能的催化作用,吡咯的结构也较文献报道的吡咯类含氮化合物结构简单,对分子结构更复杂的吡咯类含氮化合物以及地质环境中其它无机因素的考察工作拟在下一步模拟试验中进行。
动力学研究表明,吡咯生成呋喃的热化学反应的活化能为109.35 kJ/mol,除了温度、含水量以及金属氧化物以外,实际地质条件下还有很多的因素可能影响到吡咯类化合物-水-金属氧化物之间的有机-无机相互作用。这些因素包括:pH值、Eh值、有机酸及其盐类、金属离子、黏土(尤其是蒙脱石)、硅土等,这些因素对吡咯生成呋喃的热化学反应动力学的影响还有待深入研究。与实际地质条件相比,目前的模拟实验温度过高,还没有达到以自然条件来建立实验体系的阶段。下一步模拟实验需要考察上述自然条件对吡咯类含氮化合物分布的影响,特别是除了氧化铝以外的金属氧化物(金属离子)的催化作用,以进一步降低模拟试验温度。目前模拟实验所采用的模型化合物吡咯为吡咯类化合物中最简单的含氮化合物,而水及氧化铝能否与咔唑、苯并咔唑以及二苯并咔唑等分子结构更复杂的含氮化合物发生有机-无机相互作用,还有待于进一步热模拟实验研究。在今后的定量研究上,要利用实验室得到的高温短时间情况下的实验数据,建立反应动力学模型,求得动力学参数,再利用这些参数反过来估算地质条件下低温长时间情况下吡咯类化合物-水-金属氧化物反应的可能性、反应的速度、反应的转化率以及有机-无机相互作用机理等。同时利用地质实例进行进一步的验证,为探讨储层水对吡咯类化合物消耗的估计提供理论基础。
3 结论
(1)模拟实验表明,吡咯-水-氧化铝体系在350~450℃范围内可以发生反应,产物主要为呋喃与氨气。根据以往烃源岩的生烃模拟实验,在与本实验相似的高压釜体系中,350~450℃的温度条件下,烃源岩的有机质成熟度Ro值可达到1.6%~2.3%,与此相对应的地质温度大约为110~140℃,这证明了深部碳酸盐岩储层中吡咯-水-氧化铝反应体系可以发生有机氮与无机氧热化学交换作用。温度是影响吡咯生成呋喃的重要因素,随着实验温度的升高,水相的pH值、氨气含量及有机相的呋喃含量均增大,即升高温度有利于反应的进行。
(2)反应机理研究表明,水与吡咯发生了有机氮与无机氧的热化学交换作用。氧化铝固体表面Lewis酸中心很可能对吡咯生成呋喃的热化学反应过程起到了明显的催化作用,体系含水量增大将削弱氧化铝固体表面Lewis酸中心数目进而降低氧化铝催化能力。
(3)根据动力学模型计算出的活化能为109.35 kJ/mol,该反应具有较高活化能的主要原因可能是由于吡咯环状分子结构具有较高的热稳定性。除了温度、含水量以及金属氧化物以外,实际地质条件下还有很多的因素可能影响到吡咯类化合物-水-金属氧化物之间的有机-无机相互作用。这些因素可能包括:pH值、Eh值、有机酸及其盐类、金属离子、黏土(尤其是蒙脱石)、硅土等,这些因素对吡咯生成呋喃的热化学反应动力学的影响还有待深入研究。
致谢 感谢哈尔滨理工大学丁明惠副教授以及北京石油化工学院冀德坤博士在色谱-质谱分析测试方面所给予的协助。
References)
1 刘德汉,史继扬。高演化碳酸盐岩的地球化学特征和生气规律[J]。天然气地球科学,1994,2:40-41[Liu Dehan,Shi Jiyang.Geochemical characteristics and gas-producing rules from carbonate rock with high maturity[J]。Natural Gas Geoscience,1994,2:40-41]
2 郝石生,高岗,王飞宇,等。高过成熟海相烃源岩[M]。北京:石油工业出版社,1996:1-16[Hao Shisheng,Gao Gang,Wang Feiyu, et al.High and Over Matured Marine Source Rocks[M]。Beijing:Petroleum Industry Press,1996:1-16]
3 彭平安,刘大永,秦艳,等。海相碳酸盐岩烃源岩评价的有机碳下限问题[J]。地球化学,2008,37(4):415-422[Peng Ping'an,Liu Dayong,Qin Yan,et al.Low limits of organic carbon content in carbonate as oil and gas source rocks[J]。Geochimica,2008,37(4): 415-422]
4 Clegg H,Wilkes H,Oldenburg T,etal.Influence ofmaturity on carbazole and benzocarbazole distributions in crude oils and source rocks from the Sonda de Campeche,Gulf of Mexico[J]。Organic Geochemistry,1998,29(1-3):183-194
5 Li M,FowlerM G,ObermajerM,etal.Geochemical characteristicsof Middle Devonian oils in NW Alberta,Canada:possible source andmaturity effect on pyrrolic nitrogen compounds[J]。Organic Geochemistry,1999,30(9):1039-1057
6 Bakr M M Y,Wilkes H.The influence of facies and depositional environment on the occurrence and distribution of carbazoles and benzocarbazoles in crude oils:a case study from the Gulf of Suez,Egypt[J]。Organic Geochemistry,2002,33(5):561-580
7 Silliman JE,Li Maowen,Yao Huangxin,et al.Molecular distributions and geochemical implications of pyrrolic nitrogen compounds in the Permian Phosphoria Formation derived oils of Wyoming[J]。Organic Geochemistry,2002,33(5):527-544
8 李素梅,庞雄奇,黎茂稳,等。低熟油、烃源岩中含氮化合物分布规律及其地球化学意义[J]。地球化学,2002,31(1):1-7[Li Sumei,Pang Xiongqi,LiMaowen,etal.Characteristicsof pyrrolic nitrogen compounds and their geochemical significance in oils and rocks of Bamianhe oilfield,eastern China[J]。Geochimica,2002,31(1): 1-7]
9 Bennett B,Lager A。,Russell C A,et al.Hydropyrolysis of algae, bacteria,archaea and lake sediments:insights into the origin of nitrogen compounds in petroleum[J]。Organic Geochemistry,2004,35 (11-12):1427-1439
10 Zhang Chunming,Zhang Yuqing,Zhang Min,etal.Carbazole distributions in rocks from non-marine depositional environments[J]。Organic Geochemistry,2008,39(7):868-878
11 Larter SR。,Bowler B F,Li M,et al.Molecular indicators of secondary oilmigration distances[J]。Nature,1996,383(6601):593-597
12 Clegg H,Wilkes H,Horsfield B.Carbazole distributions in carbonate and clastic source rocks[J]。Geochimica et Cosmochimica Acta, 1997,61(24):5335 5345
13 Li M,Yao H,Stasiuk L D.Expulsion on pyrrolic nitrogen compound yields and distributions in Duvernay Formation petroleum source rocks in central Alberta,Canada[J]。Organic Geochemistry,1997,26 (11-12):731-744
14 Huang Haiping,Bowler B F,Zhang Zhanwen,etal.Influence ofbiodegradation on carbazole and benzocarbazole distributions in oil columns from the Liaohe Basin,NE China[J]。Organic Geochemistry, 2003,34(7):951-969
15 Snyder L R.Distribution of benzocarbazole isomers in petroleum as evidence for their biological origin[J]。Nature,1965,205-277
16 Schmitter JM,lgnatladis I,Arpion P J.Distribution of diaromatic nitrogen bases in crude oils[J]。Geochimica et Cosmochimica Acta, 1983,47(11):1975-1984
17 Dorbon M,Schmitter JM,Garrigues P,et al.Distribution of carbazole derivatives in Petroleum[J]。Organic Geochemistry,1984,7 (2):111-120
18 Patience R L,Baxby M,Bartle K D,et al.The functionality of organic nitrogen in some recent sediments from the Peru Upwelling Region[J]。Organic Geochemistry,1992,18(2):161-169
19 Baxby M,Patience R L,Bartle K.D.The origin and diagenesis of sedimentary organic nitrogen[J]。Journal of Petroleum Geology, 1994,17(2):211-230
20 Seewald JS.Aqueous geochemistry of low molecular weight hydrocarbons at elevated temperatures and pressures:Constraints from mineral buffered laboratory experiments[J]。Geochimica et Cosmochimica Acta,2001,65(10):1641-1664
21 Seewald J S.Organic-inorganic interaction in petroleum-producing sedimentary basins[J]。Nature,2003,426(6964):327-333
22 Lewan M D,Winters JC,Mcdonald JH.Generation of oil-like pyrolyzates from organic-rich shales[J]。Science,1979,203(4383): 897-899
23 Siskin M,Katritzky A R.Reactivity of organic compounds in hotwater:Geochemical and technological implications[J]。Science,1991, 254(5029):231-237
24 Helgeson H C,Knox A M,Owens C E,et al.Petroleum,oil field waters,and authigenicmineral assemblages:Are they inmetastable equilibrium in hydrocarbon reservoirs[J]?Geochimica et Cosmochimica Acta,1993,57(14):3295-3339
25 Stalker L,Farrimond P,Larter S R.Water as an oxygen source for the production of oxygenated compounds(including CO2precursors) during kerogen maturation[J]。Advanced Organic Geochemistry, 1994,22(3-5):477-486
26 Lewan M D.Experiments on the role of water in petroleum formation [J]。Geochimica et Cosmochimica Acta,1997,61(17):3691-3723
27 Mango F D,Hightower JW,James A T.Catalysis in the origin of natural gas[J]。Nature,1994,368(6471):536-538
28 Mango F D.Transition metal catalysis in the generation of natural gas [J]。Organic Geochemistry,1996,24(10-11):977-984
29 Mango FD,Hightower JW.The catalytic decomposition of petroleum into natural gas[J]。Geochimica et Cosmochimica Acta,1997,61 (24):5347-5350
30 Li Shuyuan,Guo Shaohui,Tan Xuefei.Characteristics and kinetics of catalytic degradation of immature kerogen in the presence of mineral and salt[J]。Organic Geochemistry,1998,29(5-7):1431-1439
31 李术元,林世静,郭绍辉,等。无机盐类对干酪根生烃过程的影响[J]。地球化学,2002,31(1):15-20[Li Shuyuan,Lin Shijing, Guo Shaohui,etal.Effects of inorganic salts on the hydrocarbon generation from kerogens[J]。Geochimica,2002,31(1):15-20]
32 Mackenzie A S,Patience R L,Maxwell JR,etal.Molecular parameters ofmaturation in the Toarcian Shales,Paris Basin,France.I.Change in the configuration of acyclic isoprenoid alkanes,Steranes [J]。Geochimica et Cosmochimica Acta,1980,44(11):1709-1721
33 Tannenbaum E,Kaplan IR.Role ofminerals in the thermal alteration of organic matter-I.generation of gases and condensates under dry condition[J]。Organic Geochemistry,1985,49(12):2589-2604
34 Lu Shantan,Ruth E,Kaplan IR.Pyrolysis of kerogen in the absence and presence ofmontmorillonite-I,the generation,degradation and i-somerization of steranes and triterpanes at200 and 300℃[J]。Organic Geochemistry,1989,14(5):491-499
35 冯金城。有机化合物结构分析与鉴定[M]。北京:国防工业出版社,2003:16-54[Feng Jincheng.Structure Analysis and identification of organic compounds[M]。Beijing:National Defense Industry Press,2003:16-54]
36 邢其毅,徐瑞秋,周政,等。基础有机化学[M]。北京:高等教育出版社,1994:869-879[Xing Qiyi,Xu Ruiqiu,Zhou Zheng,et al.Organic Chemistry[M]。Beijing:Higher Education Press,1994: 869-879]
37 迪安JA。兰氏化学手册[M]。北京:科学出版社,1991:9-1~9-67。[Dean JA.Lange's Handbook of Chemistry[M]。Beijing:Science Press,1991:9-1~9-67]
38 傅家谟,秦匡宗。干酪根地球化学[M]。广州:广东科技出版社, 1995:544-547[Fu Jiamo,Qin Kuangzong.Kerogen Geochemistry [M]。Guangzhou:Guangdong Science&Technology Press,1995: 544-547]
39 程克明,王兆云,钟宁宁,等。碳酸盐岩油气生成理论与实践[M]。北京:石油工业出版社,1996:1-30[Cheng Keming,Wang Zhaoyun,Zhong Ningning.Geochemistry of Source Rock[M]。Beijing:Science Press,1996:1-30]
40 朱洪法。催化剂载体制备以及应用技术[M]。北京:石油工业出版社,2002:151-152[Zhu Hongfa.Preparation of Catalyst Support and Application[M]。Beijing:Petroleum Industry Press,2002:151-152]
Study of Thermochem ical Exchange Effect between Organic Nitrogen and Inorganic Oxygen in Carbonate Rocks
DING Kang-le1WANG Hui2LUO Yue1YANG Huan1YU Qi-ming3
(1.School of Chem istry and Environmental Engineering,Yangtze University,Jingzhou,Hubei434023; 2.Daqing Oilfield Exp loration&Development Institute,Daqing,Heilongjiang 163712; 3.School of Engineering,Griffith University,QLD 4111,Australia)
The preservation of pyrrolic nitrogen compounds in deep-buried carbonate reservoirs aremost likely influenced by water and metallic oxides.In this paper,thermal simulation experiments on the pyrrole-H2O-Al2O3system were carried out using an autoclave at definite temperature and pressure.The properties of the productswere characterized by gas-chromatography and Fourier transform-infrared spectrometry methods to investigate the reaction pathway.On the basis of the experimental data,the reaction mechanism and kinetics of the pyrrole-H2O-Al2O3system were discussed tentatively.It is found that furan and ammonia were the main products during the reaction,and increasing temperature is favored.Thermochemical exchange effect between organic nitrogen and inorganic oxygen were obviously catalyzed by Al2O3,but inhibited by the increasing volume ofwater.In the presence of Al2O3,the calculated activation energy of the reaction is 109.35 kJ/mol.
carbonate reservoir;pyrrole-H2O-Al2O3system;thermal simulation experiment;reaction mechanism; kinetics
丁康乐 男 1976年出生 博士 讲师 石油与天然气化学 E-mail:dingkl2001@yahoo.com.cn
P593
A
1000-0550(2011)06-1180-10
①国家自然科学基金项目(批准号:40902034)和长江大学人才引进科研启动基金(编号:D081027)资助。
2010-11-22;收修改稿日期:2011-03-28
