Coorperativity between Metals,Ligands and Solvent:a DFT Study on the Mechanism of a Dizinc Complex-Mediated Phosphodiester Cleavage
2011-12-12FANYuBoGAOYiQin
FAN Yu-Bo GAO Yi-Qin,2,*
(1Department of Chemistry,Texas A&M University,College Station,TX 77843-3255,USA; 2College of Chemistry and Molecular Engineering,Peking University,Beijing 100871,P.R.China)
Metals in phosphate ester hydrolases play an essential role in the efficient and proficient hydrolyzation of DNA,RNA and/or other phosphate esters in cells[1-6]by catalyzing the cleavage of the exceptionally stable phosphodiester linkages[7-8].These enzymes,especially zinc-containing ones,accelerate phosphodiester hydrolysis by up to 1016-fold under neutral conditions[4-6].Zinc clusters were found at the active site in many enzymes,such as P1 nuclease[9-11]and phospholipase C[12-14],in which three Zn ions were found in the active site,and alkaline phosphatase[15-16],in which two Zn ions and one Mg ion were found.A catalytic mechanism was proposed for these enzymes,in which the two closely spaced Zn ions deprotonated the attacking water while the third one(Mg2+in alkaline phosphatase)stabilized the leaving alkoxide group.The metals are coordinated and stabilized by histidines,carboxylates(in aspartate,glutamate,or carbamate produced by CO2and lysine)and waters/hydroxides.In general, water or the 3′-OH of RNA can be deprotonated to become better nucleophiles by coordinating to Zn(II)ion(s),and therefore is able to attack phosphorus atom in the linkage to break the DNA/RNA chains.On the other hand,Zn(II)ions also play a role in binding of the phosphodiester linkages so that hydroxide/ alkoxide is oriented to attack the phosphorus atom easily.
Based on the structures and functions of the metalloenzymes, many N,O-ligands have been designed and the associated metal complexes have been synthesized to simulate the active site for phosphodiester cleavage in enzymes.Interesting catalytic activities and kinetics were reported on these metal complexes although none has nearly approached the ultimate effectiveness of the enzymes mentioned above[17-36].Among these complexes,dinuclear Zn(II)complexes have been shown to significantly accelerate the hydrolysis of phosphodiesters.
In particular,the dinuclear Zn(II)complex,shown in Scheme 1,has been reported to be an efficient catalyst for the cleavage of HPNP(HA),a model for RNA linkage(and other RNA analogues),which produces propylene phosphate(B)via transesterification in neutral or slightly basic solutions(see Scheme 1)[37-38]. This catalyst reaches its maximum catalytic activity at pH≈8.5, which is consistent with the pKa(8.0)of a Zn-bound water[37-39]. The X-ray crystal structure indicates that two zinc ions are bound together by the pendent hydroxyl on the linkage of the bis (1,4,7-triazacyclononane)ligand and,the potentiometric titrations show that the bridging hydroxyl has been deprotonated in the complex even at pH as low as 6[38].Compared to the uncatalyzed reaction(see Scheme 1),the reaction barrier for the cleavage of HA is reduced by 38.9 kJ·mol-1at pH 7.6 when this catalyst is employed[37-38].
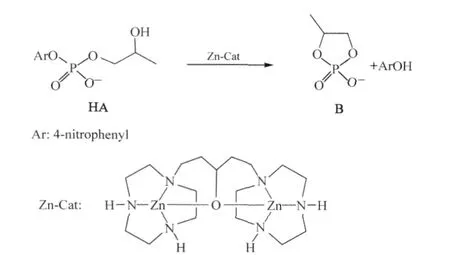
Scheme 1 Catalytic reaction and structure of dinuclear Zn(II)complex
Furthermore,the absence of a primary kinetic solvent deuterium isotope effects(SDIE)on cleavage of the substrate indicates thatproton transferisnotinvolved in the rate-determiningstep[40].
Although a large amount of thermodynamic,kinetic and X-ray crystal data have been accumulated for the phosphodiester cleavage catalyzed by dizinc complexes[37-40],the corresponding reaction mechanism at the molecular level has not been revealed.For instance,how the phosphodiester binds to the complex?Do five-coordinated phosphorus intermediates or transition states exist in the P—O forming and/or breaking processes? Are those amino protons involved in the catalysis?How many water molecules are directly involved?In this paper,these questions were tried to be answered with an extensive atomic-level investigation preformed using density functional theory(DFT).
1 Computational details
All calculations were performed using the Gaussian 03[41]implementation of Tao-Perdew-Staroverov-Scuseria(TPSS)densityfunctional theory[42].As a third generation of density functional, TPSS is generally superior to previously developed nonempirical functionals,and virtually matches in accuracy the most popular functional-B3LYP[43-46].
The basis sets for zinc and phosphorus were Stuttgart/Dresden (SDD)with the effective core potential(ECP)[47]and LANL2DZ plus a d-polarization and a p-diffuse function[48]with ECP[49-50], respectively.Basis set 6-31++G(d′,p′)was used for N,O,aromatic C,and all H atoms bound to N and O[51-53],while 6-31G was used for alkyl C and the rest of all H atoms[51].In addition, density fitting functions were included to accelerate the computation with DFT[54-55].Density fitting functions can be included for this pure DFT,and only pure DFT,to save tremendously computational expense by expanding the density in a set of atomcentered functions when computing the Coulombic interaction instead of computing all of the two-electron integrals[54-55].A comparison of TPSS with or without density fitting functions included for an organometallic reaction shows only marginal difference for both relative energies and structural parameters[56]. TPSS reproduces their properties at least as precisely as B3LYP. Especially,TPSS can recognize relatively weak interactions (such as agostic interactions)very well.Furthermore,using smaller basis sets for the atoms far from the reaction center does not degrade the computational precision and accuracy significantly but can accelerate the calculations considerably[56].
All structures were fully optimized and frequency analyses were performed to ensure that a minimum or transition state was achieved.Furthermore,transition states were confirmed with the intrinsic reaction coordinate(IRC)method[57-58]for the correct connection between the corresponding reactant and product.The thermodynamic functions,including enthalpies, entropies,and free energies,were calculated at 298.15 K and 101.325 kPa.The solvation energies for all species in aqueous solution were estimated with the polarizable conductor calculation model(CPCM)[59-60].All thermodynamic functions used for comparison and discussion as follows are from the solvation model simulations if not described with other models.
2 Results and discussion
As shown in Scheme 2,the discussion on which are the active forms of the catalyst and the substrate,is complicated by four possible combinations of these two pairs of conjugate acid-base. On the other hand,all four combinations can lead to the same binding form.For example,via a proton transfer,HA and Catform the same adduct as A-and CatH do;so do HA and CatH, if they bind first and then deprotonate.Namely,they can yield identical pH-rate profiles.A similar situation was discussed in detail in our previous published paper[61].In order to focus the investigation on the catalysis after binding,the hydroxo dizinc complex(Cat-)and the hydroxyl form of the substrate(HA) were calculated for the catalytic mechanism as they are dominating species under the optimal conditions(pH 8.5-10).
To identify the coordinating situation of the phosphate unit in the substrate,which can be a monodentate,bidentate or even tridentate ligand,the X-ray crystal structures for dizinc-phosphate complexes including a Zn-O-Zn core were thoroughly searched. The available data indicate that the phosphate bridges the zinc ions utilizing two terminal phosphate oxygens[62-67].Thus,in our calculation the phosphodiester was positioned as a bidentate ligand bridging the two zinc ions in the initial binding structure for optimizations while the hydroxyl or alkoxide on the side chain is loosely hydrogen-bonded to the Zn-bound water or hydroxide (see 1ta/1′ta:HA+Cat-and 1tb/1′tb:A-+CatH in Scheme 3).Interestingly,two amino protons are only 0.4 nm apart in the crystal structure[38]and this characteristics allows a water to be perfectly bound to the ligand by forming two hydrogen-bonds to these two amino protons.Furthermore,this water hydrogenbonds to the phenoxyl oxygen not only to orient the phosphodiester for nucleophilic attack but also to weaken the bond between the phosphorus and the phenolate for completing the reactions(see the structures in Scheme 3 and discussion below).In the case of 1′ta,the hydroxyl in substrate is spontaneously deprotonated by the Zn-bound hydroxide and exchanges with the newly-generated water to coordinate to Zn and then 1′a forms in a fully geometric optimization.This hydrogen-bonded water in 1′a can be released into the solvent to form 1a.On the other hand,in 1′tb the alkoxide can exchange with a Zn-bound water to form 1′a although 1′tb is far less stable than 1′ta because both deprotonating of the hydroxyl in substrate and protonating of the Zn-bound hydroxide are disfavored in energy in the pH range of 8.5-10.Else,with one less Zn-bound water,hydroxyl in 1ta or alkoxide in 1tb can easily coordinate to Zn and hydrogen-bond to the Zn-bound water(spontaneous proton transfer from the hydroxyl to the Zn-bound hydroxide in 1ta)to form 1a.Therefore,1a and 1′a,as monocations,were used as the starting point of the catalysis after binding.
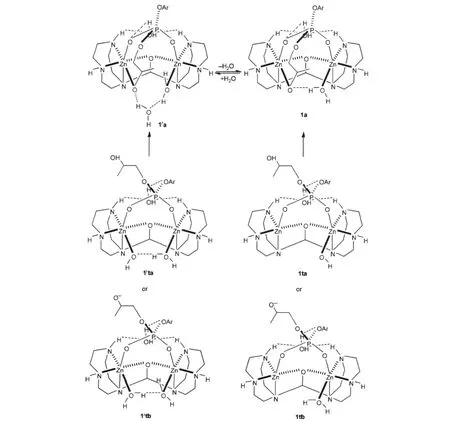
Scheme 3 Starting structures of the catalyses after binding of the catalyst to the substrate
2.1 Equilibrium between 1′a and 1a
As shown in Scheme 3,the water molecule,connecting the alkoxide on the side chain of the substrate and the Zn-bound water in 1′a,is missing and solvated into the bulky solution in 1a. 1a+H2O are 42.5 kJ·mol-1(in enthalpy)less stable than 1′a in gas phase,but when the solvation of these species and the entropy change in this equilibrium are considered,1a+H2O becomes even more stable by 55.2 kJ·mol-1than 1′a.In 1a,the hydroxyl on the side chain has been deprotonated and coordinates to Zn with a bond length of 0.2025 nm.The proton has been transferred to the original hydroxide bound to Zn to form a water molecule, which is still hydrogen-bonded to the alkoxide.The water clamped by the two amino protons with two hydrogen-bonds (O7—H1:0.2156 nm;and O7—H2:0.2169 nm)on the back (see 1a in Scheme 3 and Fig.1)has a third hydrogen-bond to one phosphate oxygen with a distance(O5—H6)of 0.1755 nm. Thus,the reaction path from 1a is more than 42 kJ·mol-1favored at the starting point than that from 1′a.
2.2 Reaction path starting with 1a
All structures involved in this path are depicted in Scheme 4 and Fig.1 and,the corresponding relative free energies are listed in Table 1.1b was also located as an isomer of 1a.Although 1b is similar to 1a,the hydrogen-bonding situation of the ligandbound water is different.Namely,the hydrogen-bond between O7 and H1 is broken with a long distance of 0.4547 nm and another hydrogen-bond from H6 to O5 has been switched to O3 with a distance of 0.1993 nm while the one between O7 and H2 has been enhanced(O—H distance of 0.1999 nm relative to 0.2169 nm in 1a).Surprisingly,these two isomers are almost isoenergetic with 1b being slightly less stable than 1a by 1.05 kJ·mol-1in free energy.The transition state between them is shown in Fig.1 as TS1,where the hydrogen-bond O7—H1 is already broken with a distance of 0.3344 nm while H6 of the ligandbound water is switching its hydrogen-bond from O5 to O3 with distances of 0.2748 and 0.2469 nm,respectively.The free energy barrier for this transition is 17.0 kJ·mol-1.
TS2 in Fig.1 is the transition state between 1b and the intermediate 2 with a five-coordinated phosphorus(trigonal bipyramidal PO5).Shown in its structure(see Fig.1),the oxyanion O6 is attacking the phosphorus and they are 0.1975 nm away while the distance between Zn1 and O6 elongates to 0.2679 nm relative to 0.2024 nm in 1b.Apparently,the Zn-bound water stabilizes the oxyanion providing a strong hydrogen-bond with a distance(O6—H5)of 0.1596 nm.On the other side,the bondlength of P—O3 increases to 0.1827 nm from 0.1688 nm in 1b. When a covalent bond is formed between O6 and P with a distance of 0.1811 nm in 2,the ligand-bound water forms a hydrogen-bond with the amino proton H1.The water,using the hydrogen-bond between O3 and H6,pulls the PO5unit to rotate.This rotation leaves Zn1 five-coordinated while Zn2 is still six-coordinated.The activation free energy for this P—O bond formation is 50.1 kJ·mol-1in gas phase but only 1.2 kJ·mol-1when solvent effect is considered(see Table 1).A charge redistribution,occurred on TS2 vs 1b,indicates that the partial positive charges on P and O2 increases by 0.2 and 0.1 unit in TS2,respectively,relative to 1b.This accumulated positive charge makes TS2 a more polarized system and as a consequence the solvation energy increases significantly(by more than 42 kJ· mol-1)in water.It is important to point out that this barrier is not expected to be lowered as significantly in enzymes because the dielectric constant for the medium in enzymes is 1-4 against 80 for water.
TS3 is the transition state for the collapse of the five-coordinated phosphorus as a result of the breaking of the bond between the phosphorus and the phenolate(see Fig.1),the length of which is 0.2063 nm(it is 0.1961 nm in 2).Because of the short distances of O3—H6 and Zn1—O3,which are 0.1771 and 0.2663 nm, respectively,not only the hydrogen-bond between the ligandbound water and the phenolate but also the five-coordinated Zn assists the bond breaking and stabilizes the transition state.In 3, two products are formed as p-nitrophenolate and propylene phosphate although they are still bound to the dizinc complex. The relative free energies of TS3 and 3 in water solution are 16.95 and-24.14 kJ·mol-1,respectively.
To further ensure validity of the results,B3LYP was also employed[68-70],combined with the same basis sets for TPSS,to calculate the single point energy(SPE)and solvation free energy upon the TPSS-optimized geometries in the path shown in Scheme 4.As listed in Table 1,the relative free energies are identical in gas phase and those in water solution are also very close for these two different density functionals.Namely,there is no method-dependent deviation in the calculations.
2.3 Reaction path starting with 1′a or 1′c
All structures involved in this reaction path are shown in Scheme 5 and the corresponding relative free energies are listed in Table 2.The reaction path from 1′a to 3′is quite similar to that from 1a to 3 except that one more water is involved(the geometries are not shown in Fig.1,but available in the Supporting Information).1′a and 1′b are two isomers for the binding of the substrate and 1′a is more stable than 1′b by 12.5 kJ·mol-1.The transition state,TS1′,connects 1′a and 1′b,is less stable than 1′a in free energy by 22.0 kJ·mol-1when solvation energy is included.The Zn-bound alkoxide,via TS2′a,with a free energy barrier of 4.7 kJ·mol-1,attacks the phosphorus to form 2′,a fivecoordinated phosphorus intermediate which is more stable than 1′b by 12.7 kJ·mol-1.At last,with the help of a Zn ion and the ligand-clamped water(on the back in 2′in Scheme 5),the bond between the phosphorus and the phenolate oxygen breaks,and the phenolate transfers to the Zn ion to form 3′.This step is almost barrierless(see Table 2)and 3′is more stable than 2′by 36.6 kJ·mol-1.

Fig.1 Optimized structures for the mechanisms shown in Schemes 4 and 5Hydrogen atoms attached to carbons are omitted for clarity.The distances are in nm.
1′c,another isomer of the substrate-binding species,the hydroxyl of which on the side chain is not deprotonated,is also lo-cated(see Scheme 5 and Fig.1).The hydroxyl forms two hydrogen-bonds with the Zn-bound water and hydroxide and 1′c is even more stable than 1′a by 5.5 kJ·mol-1(see Table 2).In 1′c, the hydroxyl is in an excellent position ready to attack the phosphorus:The hydroxyl oxygen is 0.2881 nm away from the phosphorus;the Zn-bound hydroxide tends to deprotonate the hydroxyl by forming a very strong hydrogen-bond(O9—H5: 0.1490 nm)with it;and the hydroxyl,phosphorus and the phenolate are nearly linear(O6—P—O3:166.9°)so that the formation of a five-coordinated phosphorus does not need a large conformational change.Indeed,the transition state,TS2′c,is very similar to 1′c but P—O6 is shortened to 0.2510 nm while the proton is transferring from O6 to O9(O6—H5:0.1264 nm and O9—H5:0.1180 nm in Fig.1).After the trigonal bipyramidal PO5forms,one Zn-bound water dissociates and forms hydrogenbonds with another Zn-bound water and one phosphate oxygen, leaving one Zn ion five-coordinated.The activation free energy for this step is only 4.8 kJ·mol-1.As a consequence,since the formation of 2′from 1′c is much easier than from 1′a,TS1′corresponds to the highest barrier in this reaction path.
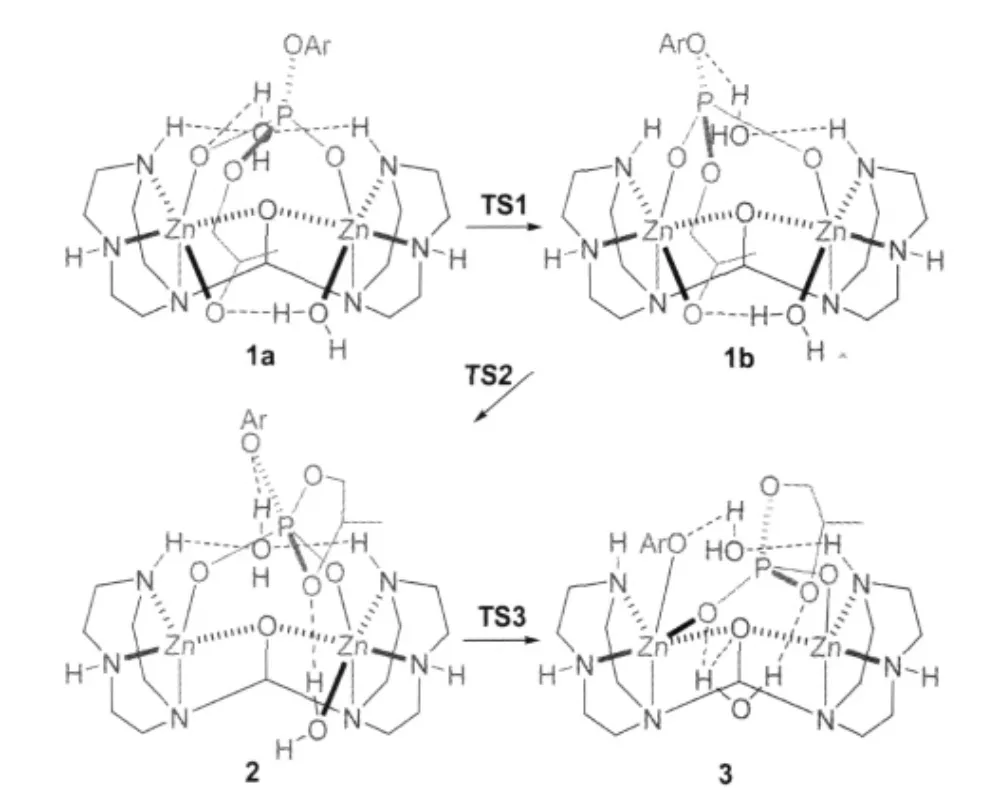
Scheme 4 Reaction mechanism starting with 1a
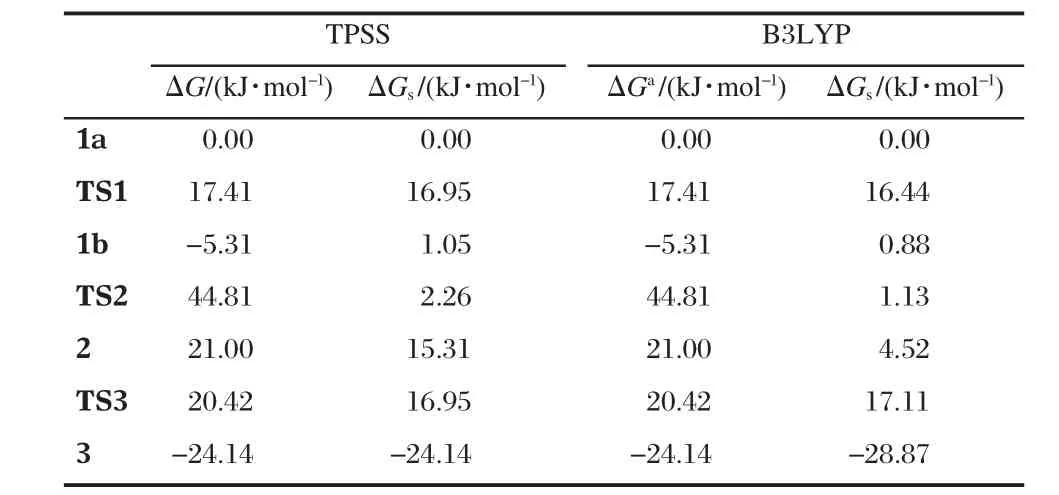
Table 1 Relative free energies in gas phase,ΔG,and in water solution,ΔGs,for the structures shown in Fig.1 involved in the reaction path in Scheme 4
2.4 Comparison of the two paths
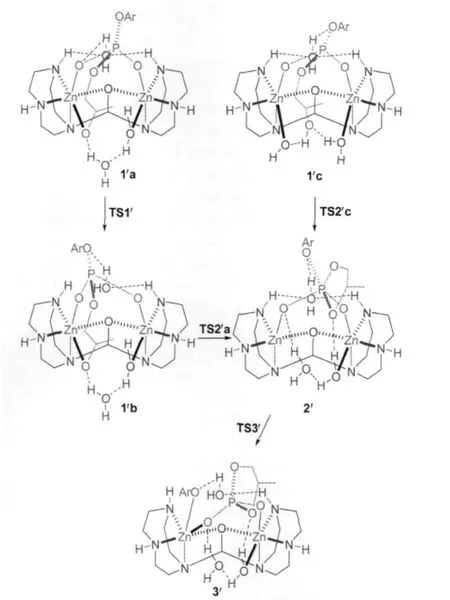
Scheme 5 Reaction mechanism starting with 1′a or 1′c
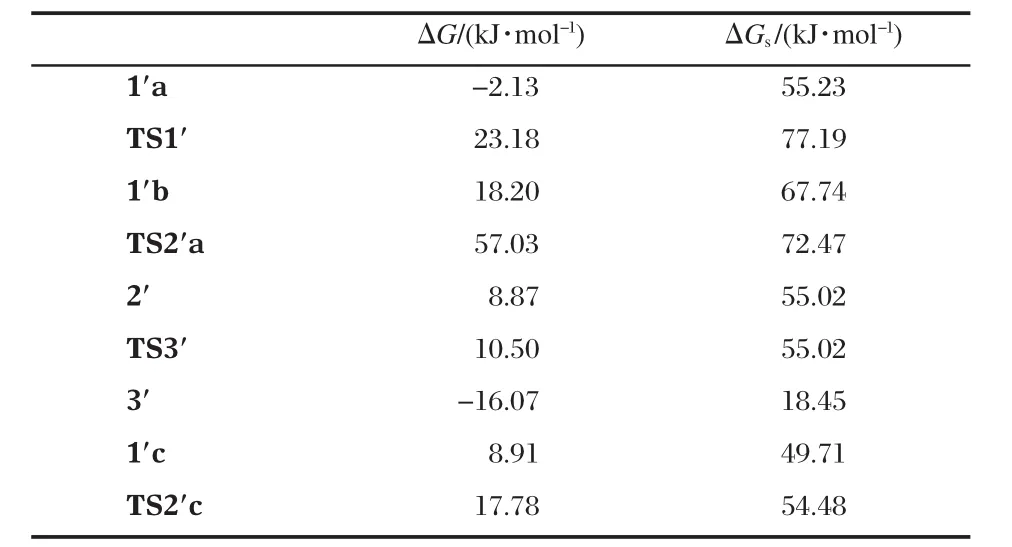
Table 2 Relative free energies in gas phase,ΔG,and in water solution,ΔGs(kJ·mol-1),for the structures shown in Fig.1 involved in the reaction path in Scheme 5
As shown in Fig.2,although the overall free energy barriers, 17.0 kJ·mol-1from 1a to 3 and 22.0 kJ·mol-1from 1′c to 3′,for the two paths shown above are similar,the fewer-water pathway(1a to 3)is favored by 55.5 kJ·mol-1when the entropy of dissociation of a water and the solvation of it into the bulky solvent are considered.Furthermore,in the path from 1′c to 3′most of the species have two six-coordinated Zn ions while in that from 1a to 3 most of them have one five-and one six-coordinated Zn ions,which is suggested by the crystal structure to be stable[38].
2.5 Rate-limiting step
The calculations show that the overall free energy barrier for the cleavage of the bound substrate is only 17.0 kJ·mol-1.The reaction rate would be too fast compared to the experiments if either the formation or the collapse of the trigonal bipyramidal PO5intermediate is the rate-determining step.The results indicate that as long as HPNP binds to the dizinc complex,it breaks down quickly to two bound products.Therefore,it is very likely that the dissociation of one product,p-nitrophenolate,is the ratedetermining step,given the increase of 1.1×107-fold(overall free energy barrier decreases by 38.9 kJ·mol-1)for the catalyzed reaction at pH 7.6 relative to the uncatalyzed reaction(actually catalyzed by OH-)[37-38],while the rate acceleration could be as large as 1016-fold in enzyme-mediated reactions.This slow release of products is likely due to its requirement of breaking a coordination bond and a hydrogen-bond simultaneously.The observation that the cleavage is faster when the leaving group is p-nitrophenolate than alkoxides is also consistent with product release being rate-determining[37].The coordination bond between Zn and p-nitrophenolate is certainly weaker than that between Zn and alkoxides.Furthermore,p-nitrophenolate can be easily solvated as an anion in neutral conditions but alkoxides are protonated[71-73].
As implied by a monozinc complex[61,74],although the PO5intermediate cannot be stabilized in the catalysis as much as by the dizinc complex,the p-nitrophenolate is loosely bound by hydrogenbonds to the Zn-coordinating water and ligand and can be easily released by exchange with the solvent or substrate.Feng et al.[75]further reported that increasing the acidity of the Zn-bound water by introducing hydrogen-bond donors could be a more effective strategy to design and to synthesize Zn catalysts than making a dinuclear analogue.It is likely that the extra hydrogenbonds not only stabilize the Zn-bound hydroxide/alkoxide as a better nucleophile but also hinder the interactions between Zn and the leaving phenolates or alkoxides.
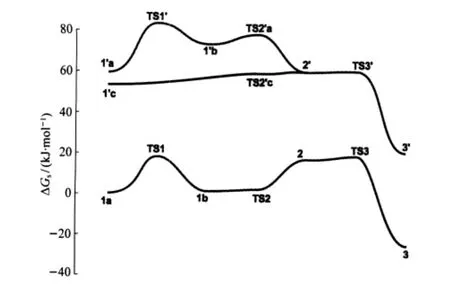
Fig.2 Relative free energies(including solvation energy)of the species for the mechanism shown in Schemes 4 and 5
In the hydrolases mentioned earlier,the strong bonding between the alkoxide and metals should be largely weakened by inhibiting the strong coordination between alkoxides and the dizinc core,which is buried relatively deep from the protein surface for DNA-binding so that the substrates are unable to interact with it directly,and forming either hydrogen-bonds or π-π stacking from the amino acid residues,which assists to pull the alkoxide(likely to be protonated to alcohol first)away,around the active site.For example,in P1 nuclease(PDB ID:1AK0)the substrate(inhibitor in the crystal)binds to the remote Zn and Arg48 and thus is unable to directly interact with the closely spaced dizinc core that is not on the surface of the enzyme.It is proposed that the dizinc center is responsible for deprotonating of the bridging water to hydroxide,which attacks the phosphate linkage and cleave it[9].The releasing of products is perfectly optimized by pulling the 3′-strand(alkoxide)away from the active center with the Phe61 stacking(π-π interaction)on the aromatic base ring in the nucleotide and Asp63 hydrogen-bonding to this base[9].
In the small dizinc complexes,weakening of the coordination bond from phenolate/alkoxide to Zn is much inferior to enzymes although the formation and collapse of the trigonal pyramidal PO5intermediate are nearly perfectly optimized.Thus,the introduction of more hydrogen-bond donors,such as amino,could lead to a better design in the developing of new catalysts,which might accelerate the releasing of phenolate/alkoxides as efficiently as enzymes.Recently,a dizinc complex,analogous to the one we calculated(see Scheme 1)but without the pendelant hydroxyl,was shown to accelerate the cleavage of HPNP by 4× 1014-fold in alcohols at pH 9.5[76-77].As reported,the weaker solvent effect of alcohols,relative to water,enhance the binding between the complex and the substrate so that the concentration of the substrate-bound catalyst increases.But consequently,the binding between the complex and the products will also be tighter to the similar extent and these two effects very likely cancel out.A more reasonable explanation might be that the entropy effect(C2H5O-or CH3O-as incoming anion)prevents the formation of a strong Zn-O-Zn core so that one Zn ion tends to bind the phosphate and the other one assists the nucleophilic attack of the hydroxyl in the substrate during the binding situation.Namely,two Zn ions act relatively independently and this flexibility increases the catalytic activity significantly.
3 Conclusions
Based on our DFT calculations,the details of the coordinations of Zn ions,the protonation and deprotonation processes, hydrogen-bonds between ligands and substrate,and collaboration between ligands,were revealed for the mechanism of the phosphodiester cleavage catalyzed by the dizinc complex.The results indicate a general base catalysis mechanism,which is consistent to the corresponding kinetics in the experiment[40].Upon binding,the substrate bridges two Zn ions by forming two coordination bonds between two terminal phosphate oxygens and two zinc ions.The hydroxyl on the side chain is flexible to coordinate to the Zn ion and can be easily deprotonated by the Znbound hydroxide.A finding on the delicacy of the catalyst is that two amino protons are close to perfectly bind a water molecule, which is able to position the substrate in a good orientation not onlyforthe nucleophilic attack(to formthe five-coordinated phosphorus intermediate)but also for the collapse of the intermedi-ate to the products.The flexibility of Zn ion′s coordination(5 or 6)assists the formation and breaking of P—O bonds and significantly stabilizes the corresponding transition states.The Znbound water stabilizes the oxyanion in the nucleophilic attack by forming a strong hydrogen-bond while the ligand-bond water lowers the energy for the breaking the P—O bond between the phosphorus and the phenolate.
The overall activation free energy,during the binding situation,is only about 17.0 kJ·mol-1,which is too low to be the ratedetermining step comparable to the experiments.Based on these results,we conclude that the slowest step in the complete catalytic cycle very likely is the releasing of one product,p-nitrophenolate because it is quite difficult to break the strong coordination bond between Zn and the phenolate and related hydrogenbonds simultaneously.
Supporting Information Available: Cartesian coordinates,electronic energies,zero-point energies,enthalpies,free energies,and solvation free energies for all species included in Figs.(1,2)and complete Ref.[41]have been included.This information is available free of charge via the internet at http:// www.whxb.pku.edu.cn.
1 Weston,J.Chem.Rev.,2005,105:2151
2 Jedrzejas,M.J.;Setlow,P.Chem.Rev.,2001,101:607
3 Cowan,J.A.Chem.Rev.,1998,98:1067
4 Wilcox,D.E.Chem.Rev.,1996,96:2435
5 Strater,N.;Lipscomb,W.N.;Klabunde,T.;Krebs,B.Angew. Chem.Int.Edit.,1996,35:2024
6 Lipscomb,W.N.;Strater,N.Chem.Rev.,1996,96:2375
7 Schroeder,G.K.;Lad,C.;Wyman,P.;Williams,N.H.; Wolfenden,R.Proc.Natl.Acad.Sci.U.S.A.,2006,103:4052
8 Williams,N.H.;Wyman,P.Chem.Commun.,2001:1268
9 Romier,C.;Dominguez,R.;Lahm,A.;Dahl,O.;Suck,D.Proteins, 1998,32:414
10 Volbeda,A.;Lahm,A.;Sakiyama,F.;Suck,D.EMBO J.,1991, 10:1607
11 Lahm,A.;Volbeda,A.;Suck,D.J.Mol.Biol.,1990,215:207
12 Hansen,S.;Hansen,L.K.;Hough,E.J.Mol.Biol.,1993,231:870
13 Hansen,S.;Kristian,L.;Hough,H.;Hough,E.J.Mol.Biol.,1992, 225:543
14 Hough,E.;Hansen,L.K.;Birknes,B.;Jynge,K.;Hansen,S.; Hordvik,A.;Little,C.;Dodson,E.;Derewenda,Z.Nature,1989, 338:357
15 Holtz,K.M.;Kantrowitz,E.R.FEBS Lett.,1999,462:7
16 Kim,E.E.;Wyckoff,H.W.J.Mol.Biol.,1991,218:449
17 Fritsky,I.O.;Ott,R.;Pritzkow,H.;Kramer,R.Inorg.Chim.Acta, 2003,346:111
18 Bonora,G.M.;Drioli,S.;Felluga,F.;Mancin,F.;Rossi,P.; Scrimin,P.;Tecilla,P.Tetrahedron Lett.,2003,44:535
19 Gajda,T.;Jancso,A.;Mikkola,S.;Lonnberg,H.;Sirges,H. J.Chem.Soc.Dalton Trans.,2002:1757
20 Albedyhl,S.;Schnieders,D.;Jancso,A.;Gajda,T.;Krebs,B.Eur. J.Inorg.Chem.,2002:1400
21 Fritsky,I.O.;Ott,R.;Pritzkow,H.;Kramer,R.Chem.-Eur.J., 2001,7:1221
22 Albedyhl,S.;Averbuch-Pouchot,M.T.;Belle,C.;Krebs,B.; Pierre,J.L.;Saint-Aman,E.;Torelli,S.Eur.J.Inorg.Chem., 2001:1457
23 Rossi,P.;Felluga,F.;Tecilla,P.;Formaggio,F.;Crisma,M.; Toniolo,C.;Scrimin,P.Biopolymers,2000,55:496
24 Gajda,T.;Kramer,R.;Jancso,A.Eur.J.Inorg.Chem.,2000:1635
25 Williams,N.H.;Takasaki,B.;Wall,M.;Chin,J.Acc.Chem.Res., 1999,32:485
26 Rossi,P.;Felluga,F.;Tecilla,P.;Formaggio,F.;Crisma,M.; Toniolo,C.;Scrimin,P.J.Am.Chem.Soc.,1999,121:6948
27 Blasko,A.;Bruice,T.C.Acc.Chem.Res.,1999,32:475
28 Williams,N.H.;Cheung,W.;Chin,J.J.Am.Chem.Soc.,1998, 120:8079
29 Yashiro,M.;Ishikubo,A.;Komiyama,M.Chem.Commun.,1997: 83
30 Liu,S.H.;Hamilton,A.D.Tetrahedron Lett.,1997,38:1107
31 Liu,S.H.;Hamilton,A.D.Bioorg.Med.Chem.Lett.,1997,7: 1779
32 Young,M.J.;Wahnon,D.;Hynes,R.C.;Chin,J.J.Am.Chem. Soc.,1995,117:9441
33 Young,M.J.;Chin,J.J.Am.Chem.Soc.,1995,117:10577
34 Yashiro,M.;Ishikubo,A.;Komiyama,M.J.Chem.Soc.Chem. Commun.,1995:1793
35 Chapman,W.H.;Breslow,R.J.Am.Chem.Soc.,1995,117:5462
36 Wall,M.;Hynes,R.C.;Chin,J.Angew.Chem.Int.Edit.,1993, 32:1633
37 O′Donoghue,A.;Pyun,S.Y.;Yang,M.Y.;Morrow,J.R.; Richard,J.P.J.Am.Chem.Soc.,2006,128:1615
38 Iranzo,O.;Kovalevsky,A.Y.;Morrow,J.R.;Richard,J.P.J.Am. Chem.Soc.,2003,125:1988
39 Iranzo,O.;Elmer,T.;Richard,J.P.;Morrow,J.R.Inorg.Chem., 2003,42:7737
40 Yang,M.Y.;Iranzo,O.;Richard,J.P.;Morrow,J.R.J.Am.Chem. Soc.,2005,127:1064
41 Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian 03. Revision C.02.Wallingford,CT:Gaussian Inc.,2004
42 Tao,J.M.;Perdew,J.P.;Staroverov,V.N.;Scuseria,G.E.Phys. Rev.Lett.,2003,91:146401/1
43 Staroverov,V.N.;Scuseria,G.E.;Tao,J.M.;Perdew,J.P. J.Chem.Phys.,2003,119:12129
44 Csonka,G.I.;Ruzsinszky,A.;Tao,J.M.;Perdew,J.P.Int.J. Quantum Chem.,2005,101:506
45 Perdew,J.P.;Ruzsinszky,A.;Tao,J.M.;Staroverov,V.N.; Scuseria,G.E.;Csonka,G.I.J.Chem.Phys.,2005,123:062201/1
46 Furche,F.;Perdew,J.P.J.Chem.Phys.,2006,124:044103/1
47 Dolg,M.;Wedig,U.;Stoll,H.;Preuss,H.J.Chem.Phys.,1987, 86:866
48 Check,C.E.;Faust,T.O.;Bailey,J.M.;Wright,B.J.;Gilbert,T. M.;Sunderlin,L.S.J.Phys.Chem.A,2001,105:8111
49 Hay,P.J.;Wadt,W.R.J.Chem.Phys.,1985,82:270
50 Hay,P.J.;Wadt,W.R.J.Chem.Phys.,1985,82:299
51 Hehre,W.J.;Ditchfie,R.;Pople,J.A.J.Chem.Phys.,1972,56: 2257
52 Krishnan,R.;Binkley,J.S.;Seeger,R.;Pople,J.A.J.Chem.Phys., 1980,72:650
53 Clark,T.;Chandrasekhar,J.;Spitznagel,G.W.;Schleyer,P.V. J.Comput.Chem.,1983,4:294
54 Dunlap,B.I.J.Chem.Phys.,1983,78:3140
55 Dunlap,B.I.J.Mol.Struct.-Theochem,2000,529:37
56 Fan,Y.B.;Hall,M.B.Organometallics,2005,24:3827
57 Gonzalez,C.;Schlegel,H.B.J.Chem.Phys.,1989,90:2154
58 Gonzalez,C.;Schlegel,H.B.J.Phys.Chem.,1990,94:5523
59 Barone,V.;Cossi,M.J.Phys.Chem.A,1998,102:1995
60 Cossi,M.;Rega,N.;Scalmani,G.;Barone,V.J.Comput.Chem., 2003,24:669
61 Fan,Y.B.;Gao,Y.Q.J.Am.Chem.Soc.,2007,129:905
62 Feng,G.Q.;Natale,D.;Prabaharan,R.;Mareque-Rivas,J.C.; Williams,N.H.Angew.Chem.Int.Edit.,2006,45:7056
63 Fan,H.;Slebodnick,C.;Hanson,B.E.Inorg.Chem.Commun., 2006,9:103
64 Venegas-Yazigi,D.;Cubillos,M.;Le Fur,E.;Pivan,J.Y.;Garland, M.T.;Baggio,R.;Spodine,E.Cryst.Growth Des.,2005,5:1695
65 Kinoshita,E.;Takahashi,M.;Takeda,H.;Shiro,M.;Koike,T. Dalton Trans.,2004:1189
66 He,C.;Lippard,S.J.J.Am.Chem.Soc.,2000,122:184
67 Lugmair,C.G.;Tilley,T.D.;Rheingold,A.L.Chem.Mater., 1997,9:339
68 Becke,A.D.Phys.Rev.A,1988,38:3098
69 Lee,C.T.;Yang,W.T.;Parr,R.G.Phys.Rev.B,1988,37:785
70 Becke,A.D.J.Chem.Phys.,1993,98:1372
71 Lonnberg,H.;Stromberg,R.;Williams,A.Org.Biomol.Chem., 2004,2:2165
72 Mikkola,S.;Stenman,E.;Nurmi,K.;Yousefi-Salakdeh,E.; Stromberg,R.;Lonnberg,H.J.Chem.Soc.Perkin Trans.2,1999: 1619
73 Komiyama,M.;Matsumoto,Y.;Takahashi,H.;Shiiba,T.;Tsuzuki, H.;Yajima,H.;Yashiro,M.;Sumaoka,J.J.Chem.Soc.Perkin Trans.2,1998:691
74 Feng,G.Q.;Mareque-Rivas,J.C.;de Rosales,R.T.M.;Williams, N.H.J.Am.Chem.Soc.,2005,127:13470
75 Feng,G.Q.;Mareque-Rivas,J.C.;Williams,N.H.Chem. Commun.,2006:1845
76 Wang,Q.;Lonnberg,H.J.Am.Chem.Soc.,2006,128:10716
77 Liu,C.T.;Neverov,A.A.;Brown,R.S.Inorg.Chem.,2007,46: 1778
