Al-Li-Zr合金的界面原子成键与力学性能
2011-11-24高英俊文春丽莫其逢罗志荣黄创高
高英俊, 文春丽, 莫其逢, 罗志荣, 黄创高
Al-Li-Zr合金的界面原子成键与力学性能
高英俊, 文春丽, 莫其逢, 罗志荣, 黄创高
(广西大学 物理科学与工程技术学院,南宁 530004 )
应用固体经验电子理论计算Al-Li-Zr合金中若干析出相与基体的界面原子成键强度和异相界面的界面能。结果表明,δ′相与基体之间的界面电子密度在较低的应力下保持连续,使得 δ′相与基体界面的结合较好,起到界面增强的效果;δ相与基体间界面电子密度在一级近似下不连续,使得与基体间界面结合强度较弱,引起界面结合弱化。对于核壳结构的复合相δ′/β′,界面电子密度差较小,且界面能最低,使得δ′相容易在β′相上异质形核长大形成复合δ′/β′相。由此从界面原子成键角度揭示析出相对合金起强弱化作用的原因,及其对合金力学性能的影响。
Al-Li-Zr合金;原子成键;相界面;力学性能
Al-Li合金具有低密度、高比强度和比模量,良好的耐蚀性和优异的低温特性等特点[1−3],已成为航空航天的重要结构材料。Al-Li合金中加入微量的Zr元素,能够起到晶粒细化作用,提高合金的力学性能。实验研究[4]还表明,Al-Li合金所具有的高弹性模量与 Li原子及其周围的原子成键有着紧密的关系,同时,合金的性能还与析出相界面原子成键有密切关系[5]。现在,人们更注重从微观原子键络结构揭示合金具有的优良宏观性能的内在原因[3−5]。此前,高英俊等已从原子成键的角度对 Al-Li-Zr合金的析出相[6−7],如 δ′(Al3Li)相,β′ (Al3Zr)相的价电子结构进行计算,分析了 δ′ 对合金作用机理,以及 β′ 粒子对 δ′ (Al3Li)相晶粒的细化作用的原因,而本文作者则从这些析出相的界面电子成键角度,研究析出相对合金的力学性能的影响,揭示其对合金强弱化作用的内在原因,为合金的改性设计提供理论指导。
1 结构模型
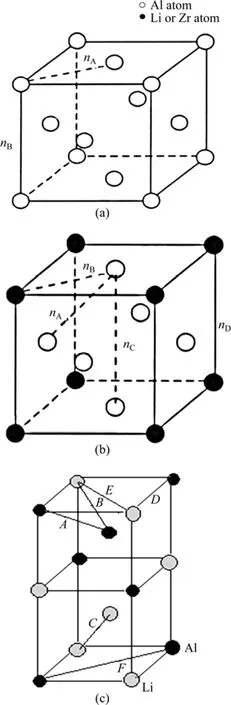
图1 晶胞结构Fig.1 Cell structure: (a) Pure Al cell; (b) Al3Li or Al3Zr cell;(c) 1/4 δ(AlLi) cell
Al-Li合金的析出相有亚稳δ′相,稳定平衡δ相。对于Al-4.2%Li合金(质量分数),已知Al晶格常数为a0=0.404 96 nm。按照文献[8]指出,铝的点阵常数随Li的增加而下降,含 4.2%Li的合金的晶格常数为 a=0.404 6 nm,其晶胞结构如图1(a)所示。δ′(Al3Li)相是面心立方L12结构,晶格常数为a=0.401 nm,属于立方晶系,具有Pm3m空间群。该相与基体完全共格,且具有(111)δ′//(111)α(Al)的位向关系,其晶体结构[8]如图1(b)所示。时效过程的最终平衡相为δ(AlLi)相,其为体心立方结构,晶格常数[8]为a=0.636 nm,属于Fd3m空间群,该相与母相非共格,与母相间的位向关系为(100)δ//(110)α(Al)。由于 δ相晶体结构的对称性,为了便于研究,采用δ相晶胞的1/4结构进行计算,其结构如图1(c)所示。
对于加入微量Zr的Al-Li合金,在过饱和固溶体中析出 β′(Al3Zr)相粒子,其晶体结构与 δ′相的结构相同[7],都是面心立方L12结构,Al原子位于晶胞结构的面心位置。Zr原子位于立方体顶角处,β′相的晶格常数是 a=0.405 0 nm,与母相间的位向关系[8]为(111)Al3Zr//(111)α(Al)。
实验观察到析出的β′相粒子可以作为δ′相的异质形核位置,形成具有核壳结构的复合双相 δ′/β′的形貌[9],δ′相的{002}面与 β′相的{001}面具有相同的原子排列,核壳结构的复合沉淀相 δ′/β′的界面取向关系为(002)δ′//(001)β′。在高分辨透射电镜下观察[9],δ′相包覆在β′相上,将β′相包围在中心,在电镜下呈现出中心暗、外围亮的球状,该复合相要比δ′相粗大得多。
2 方法与结果
2.1 EET理论简介
固体中原子的价电子结构在这里是指该固体中原子所处的状态以及原子形成共价键的键络分布。按照EET[10−11]理论,原子的共价电子是分布在连接最近邻、次近邻, 以及 s近邻原子的键上。各键上共价电子对数(即键级nα)由下列原子键距公式表示
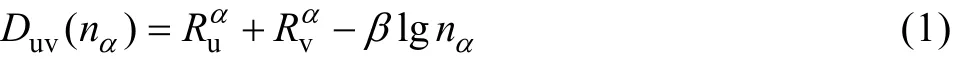
式中:Duv是共价键距;uRα和vRα是单键半距;β为参量。计算中参数β的数值按文献[10−11]中的公式确定。晶胞内的共价电子数满足下述方程关系:

式中:k1和k2分别为晶胞中 u和v原子的个数,ncu和nv分别为u和v原子的共价电子数。Iα为nα键级的等
c同键数,各等同键数的确定可依照文献[10, 12]给出的方法计算得到。由于各晶胞的结构已确定,实验晶格常数文献[8]已给出,因此,运用键距差(BLD)方法[10]建立最强键nA方程,并参见文献[6, 7, 12−14]的求解步骤,联立式(1)和(2)等方程组,逐个计算各晶胞中原子成键的价电子结构, 并依据BLD判据确定原子的杂阶状态ε。δ′、β′和δ相结构的原子共价键强结果已在高英俊等的前期研究阶段发表,详细见文献[7]中表1、4和表5,以及文献[15]中表3−2。
2.2 改进的TFD理论简介
文献[11]定义了异相界面电子结构,指出异相界面电子结构除包括相界面两侧平面上的键络电子分布外,还包括相界面两侧平面上的平均共价电子密度ρ(hkl)、ρ(uvw) 和电子密度的相对差值 Δρ,以及使界面电子密度在一级近似下保持连续的原子状态组数σ。相界面电子结构的计算在空间电子结构计算的基础上进行。
对于(hkl)α//(uvw)β异相界面电子结构的计算,首先要求出在 α和 β相空间中,符合键距差判别条件|ΔDna|≤0.05 nm的原子状态,在此基础上,应用“界面上的电子密度连续”的边界条件(在一级近似下,以Δρ<10%来判断界面电子密度的连续性。当Δρ<10%时,把电子密度定义为连续或连续性较好;当Δρ>10%时,界面电子密度认为是偏离连续或连续性较差),得到异相界面的原子键络的强度分布。然后计算该异相界面两侧平面(hkl)和(uvw)上的平均共价电子密度 ρ(hkl)和ρ(uvw)以及界面处电子密度的相对差值 Δρ,其具体计算表达式由改进的TFD理论[11]给出如下:
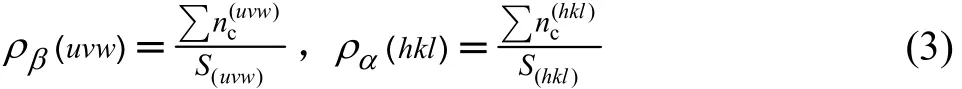
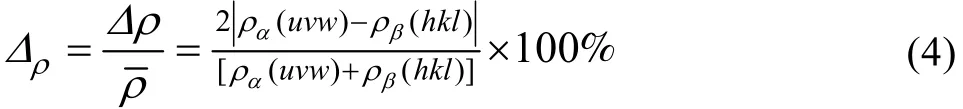
式中:nc为共价电子数; Σ n(uvw)为(uvw)或(hkl)面参考单元上的共价电子总数, Σ n(uvw)=nAIA+nBIB+c nCIC+…;S(uvw)和 S(hkl)分别为(uvw)和(hkl)面参考单元的面积;Δρ为界面处电子密度的相对差值;Δρ为面电荷密度差;ρ为平均面电荷密度;σ为满足Δρ<10%的组合数,即异相界面连续的状态数。计算得到的各异相界面的原子键络强度结果列在表 1~4,表中 Εb为键结合能[10]。
改进的 TFD理论给出的异相界面电子结构的物理意义如下:相界面处电子密度ρ愈高,界面的原子键络就越密,界面结合得就越牢固;相界面处的电子密度差Δρ愈小,界面上的电子密度连续性就愈好,界面原子键络匹配得就越好,界面畸变能就越低,界面畸变应力也愈小;反之,界面畸变应力愈大,界面畸变能就越高,界面就越不稳定。当畸变应力大到临界值时,则电子密度的连续性遭到破坏,将伴随在界面新相的生成或在宏观上出现裂纹或断裂。电子密度的连续性的好坏实质上是由于点阵原子键络畸变和缺陷而导致的结果,直接影响到材料的性能好坏。
2.3 异相界面能计算方法
2.3.1 (111)δ′//(111)α(Al)的界面能
δ′相与基体具有(111)δ′//(111)α(Al)的位向关系;δ相与母相的位向关系为(100)δ//(110)α(Al)。在上节中介绍计算了δ′和δ相与基体之间界面的电子结构的方法。由于界面能与形成界面时原子键络有关,下面介绍利用表1中δ′/α(Al)界面处的原子成键的计算结果,应用推广的Becker模型[16]来研究 δ′/α(Al)界面处的界面能。
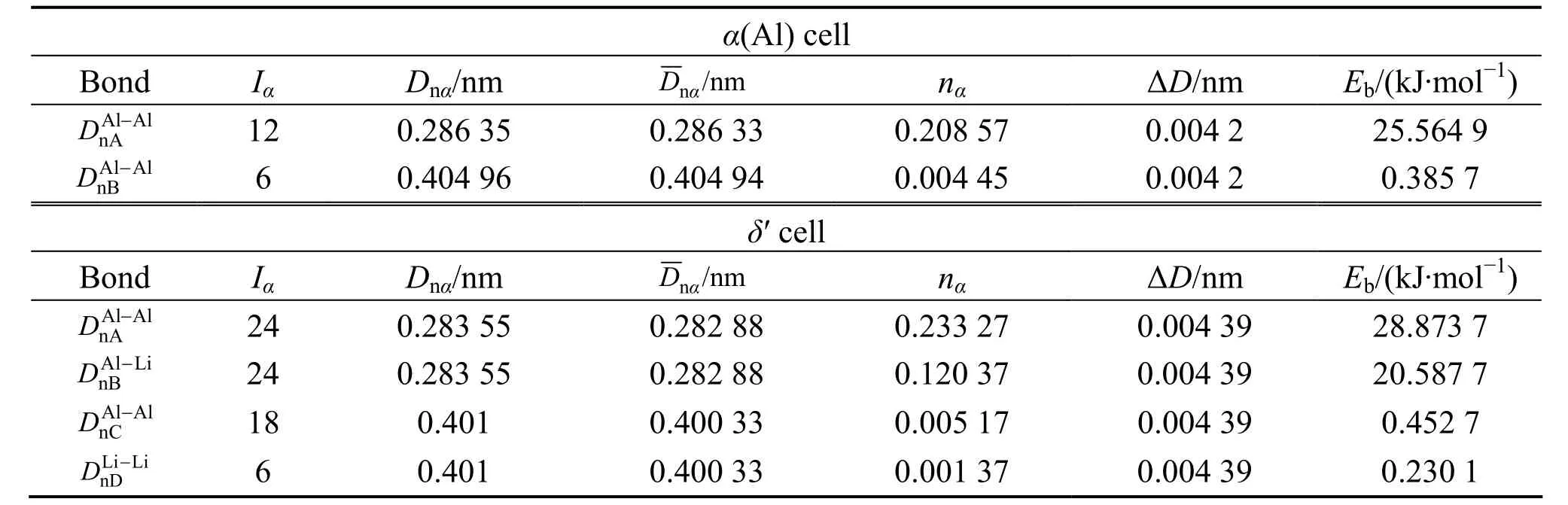
表1 (111)α(Al)/(111)δ′(Al3Li)界面原子成键Table 1 Atomic bonding of interface of (111)α(Al)/(111)δ′(AlLi)
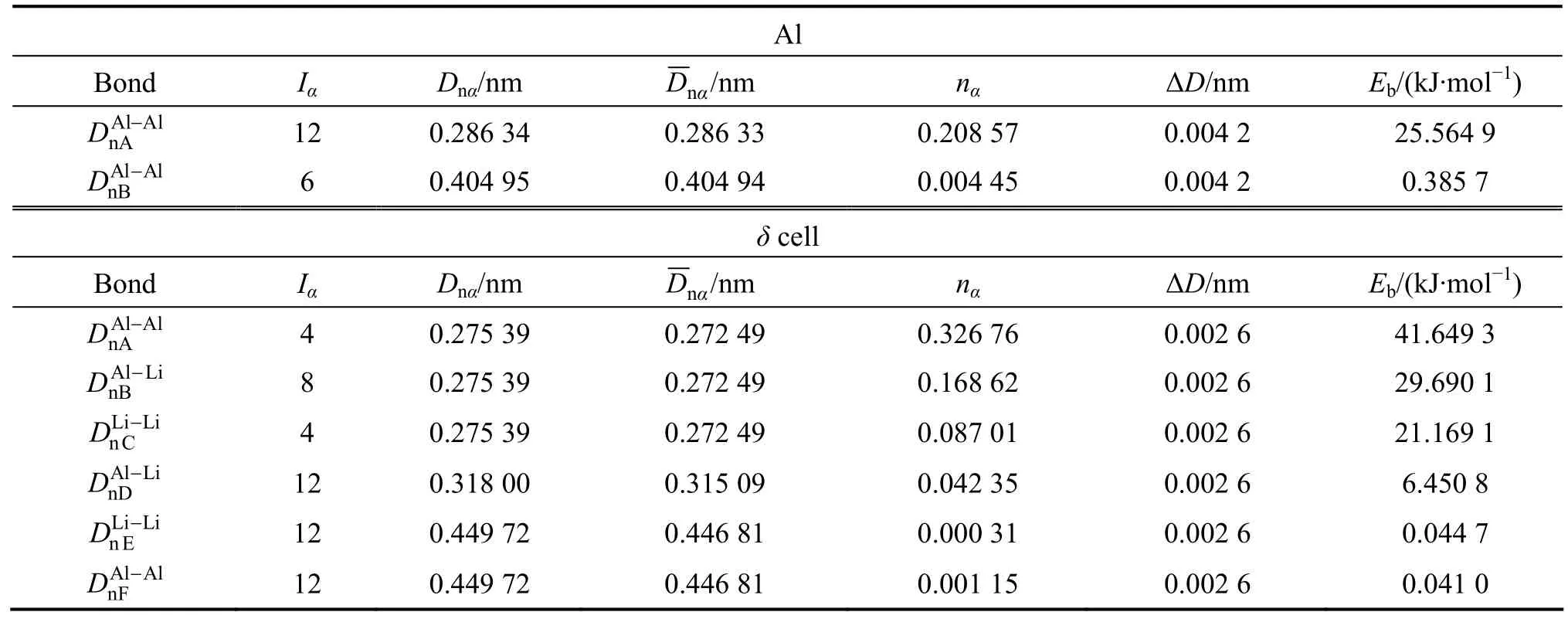
表2 (100)δ//(110)α(Al) 界面原子成键Table 2 Atomic bonding of interface of (100)δ//(110)α(Al)
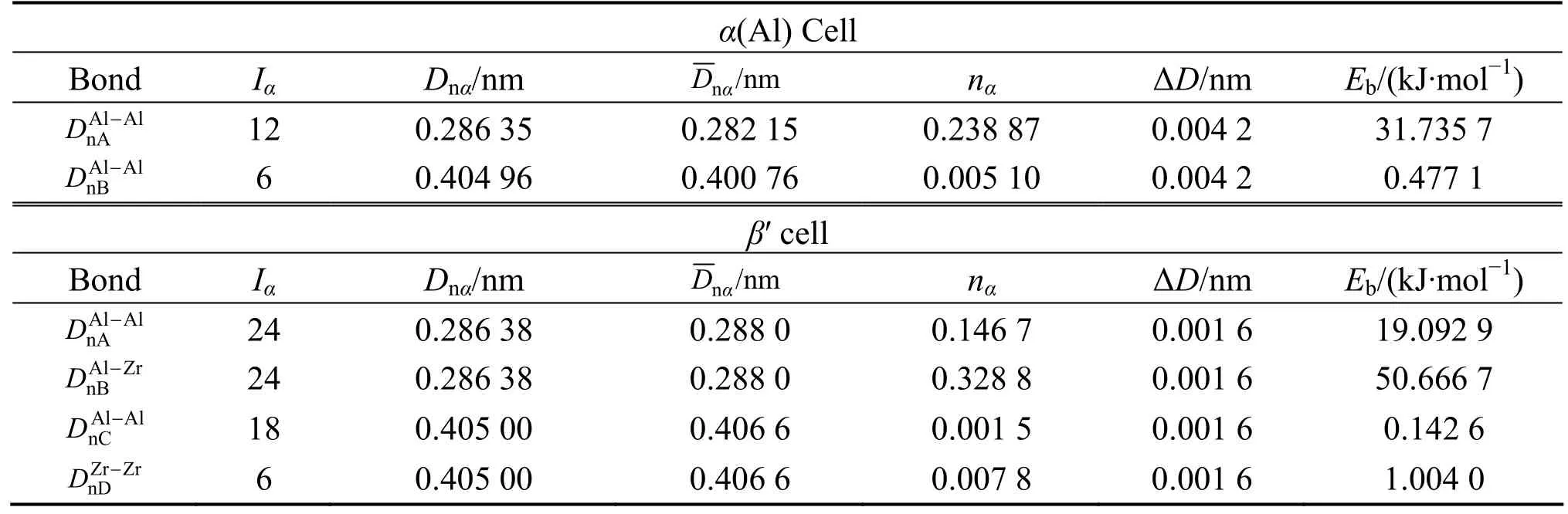
表3 (111)α(Al)/(111)β′(Al3Zr)界面原子成键Table 3 Interfacial atomic bonding of (111)α(Al)/(111)β′(Al3Zr)
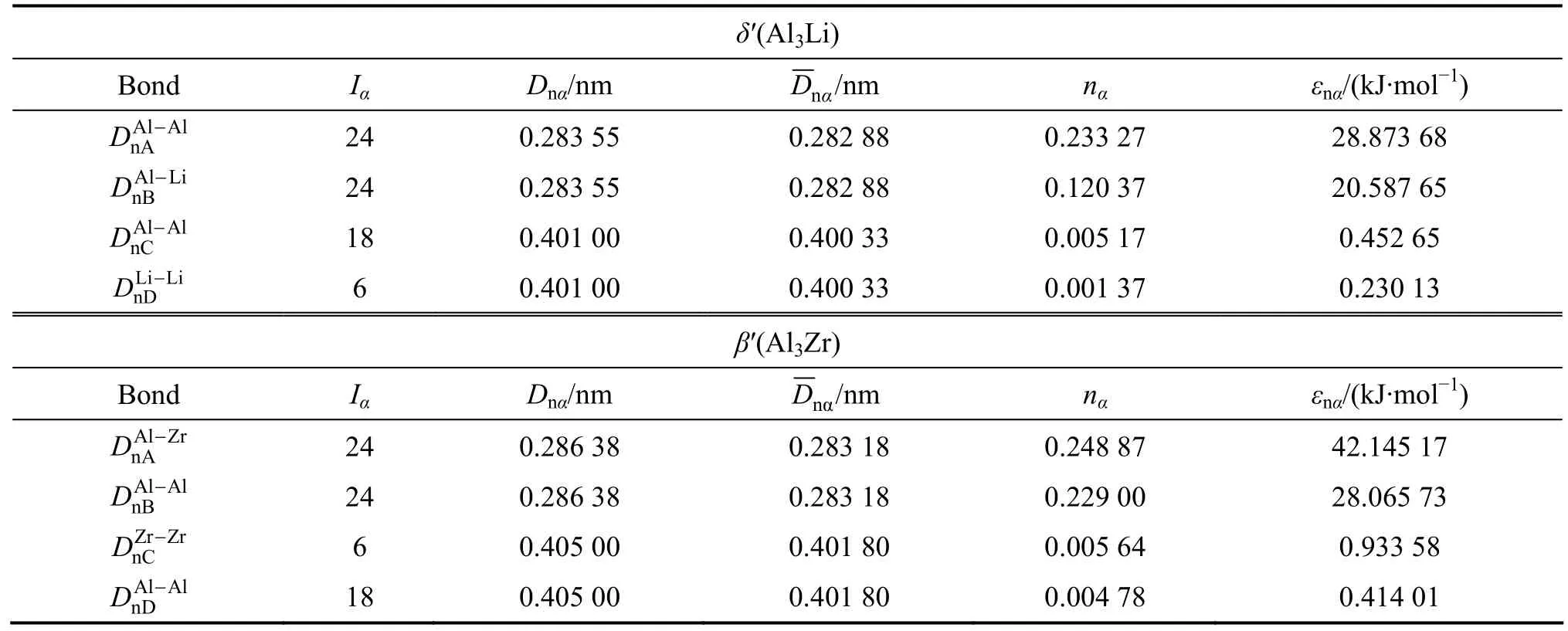
表4 (002)δ′//(001)β′界面原子成键Table 4 Interfacial atomic bonding of (002)δ′// (001)β′
根据推广的Becker模型,把基体记为α相, δ′相记为β相,把Al原子记为A原子,Li原子记为B原子,则 α(Al)/δ′界面属于 α(A-B)//β(A-B)类型的界面,推导出界面能的计算公式[16]如式(5)给出:

式中:Zint为界面上的平均键密度,等于界面上的原子密度NS与面配位数ZR的乘积,可用穿过界面的键数与界面面积的比值来计算;Δc为界面两侧溶质的浓度差; cBβ为β相中B原子的浓度;分别为α、β相中AB原子组成的键的键能;分别为α、β相中AA原子组成的键的键能;为α和β相中BB原子组成的键的键能。在基体α(Al)中,由于Li原子的浓度很小,基体中Li原子的浓度近似为6%,Al—Li键的键能近似按Al-6.25%Li固溶体中计算,根据文献[10]的计算方法,计算出 δ′相中异类原子的Al—Li键的键能为 εβ=18.122 9 kJ/mol。
对于(111)δ′//(111)α(Al)界面,4.309×1019m−2;将=4.309×1019m−2、Δc=0.25以及表 2中的各键能值代入上面式(5) 计算可得,γδ′/Al= 13.80 mJ/m2,与实验值[17]γδE'/Al= 14 mJ/m2吻合,结果列在表5。对于其它析出相与基体的界面能,如(100)δ//(110)α(Al)和(111)Al3Zr//(111)α(Al)界面,应用同样的方法计算出界面能,结果见表5。
2.3.2 δ′(002)//β′(001)界面的界面能
δ′相与 β′相的界面取向关系为(002)δ′//(001)β′,根据推广的Becker模型[16],把形成界面的两个相分别记为 α相和 β相,计算时把δ′相记为α相,β′相记为β相,把Al、Li、Zr原子分别记为A、B、C原子,则δ′//β′界面属于 α(A-B)//β(A-C)类型的界面,这时,异相界面能的计算公式[15]为
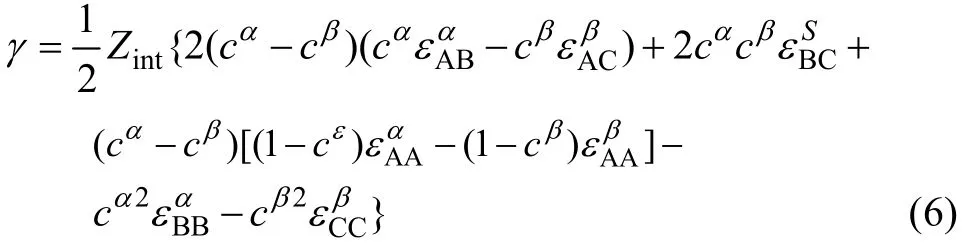
在 δ′(002)//β′(001) 界 面 处 , 计 算 得 列=4.975×1019m−2,,界面上的平均键密度按两者的平均值计算,即Zint=Zint=3.707×1019m−2;溶质的浓度近似按摩尔分数计算,即;由于界面取向关系为(002)δ′//(001)β′,δ′相与 β′相的晶格常数接近,错配度小于1%,在界面上Li、Zr原子占据相同的位置概率相等,因此,近似将取为0。将上述各值及表4中的相应键能值代入式(6),计算得到 δ′/β′界面的界面能结果列在表5中。与实验测量结果范围一致。
界面原子成键结果见表 1~4,界面能计算结果见表5。
3 分析与讨论
从表1和表2中的计算结果可以看出,对于δ′相与基体 α(A l)之间的界面,最小电子密度差Δρmin=2.4754%,表明δ′相与基体之间的界面电子密度(一级近似Δρ<10%)保持连续;对于 δ相与基体之间的界面,Δρmin=74.74%,表明δ相与基体间界面电子密度在一级近似下不连续,界面应力较大,界面结合强度较弱。另一方面,在δ′相界面处平均电子密度为17.373 nm−2,而在δ相界面处的平均电子密度为2.67 nm−2。这说明δ′相与基体界面的结合强度大,而δ相与基体间界面结合很弱,δ′相的析出起到了界面增强的效果,而δ相则引起界面强度弱化。从界面能的计算结果看出,δ′相与基体间界面能很低,只有 13.80 mJ/m2,与实验值14 mJ/m2[17]符合,说明应用推广的Becker模型计算界面能是合理的。同时,根据界面能的大小反映形核的难易程度。界面能很低说明共格δ′相在基体中极易形核,而且析出相分布大多是比较均匀的,所以析出的δ′相均匀细小。由于其Al—Li共价键络比基体中的Al—Al键络强得多,细小的δ′相均匀分布在基体中,能够有效地阻碍位错切割,对合金起到弥散强化作用,因而提高了合金的强度性能。δ相与基体间的界面能很大,近似为235.97 mJ/m2,而界面电荷连续性较差,Δρmin=74.74%。在合金的时效初期,δ相不易形核和生长,在时效后期析出δ相。由计算结果看出,δ相与基体界面结合较弱,导致δ相的析出弱化了合金的强度。总的来说,δ′相与基体间界面应力小,界面结合较强,界面稳定性好,δ′相的析出增强了合金的强度;δ相与基体相界面的界面应力很大,界面结合较弱,界面稳定性差,δ相使合金强度显著下降。因此,在Al-Li合金中,δ′相对合金起强化作用,而δ相的大量析出使合金的强度下降。

表5 Al-Li-Zr合金析出相与基体间的界面能Table 5 Interfacial energy between precipitation and matrix of Al-Li-Zr alloys
由表3的结果可见,在Al3Zr//α(Al)界面处,与基体之间的界面电子密度差较小,Δρmin=0.492 3%,说明Al3Zr与基体界面处,界面电荷连续性好,界面应力较小,界面结合得较好。Al3Zr/α(Al)界面能的计算结果为76.47 mJ/m2,与实验值[18]66.3 mJ/m2符合较好。对于核壳结构的复合相δ′/β′, 由表4可见,界面电子密度差较小(Δρmin=4.37%),其界面能最小,只有2.24 mJ/m2,说明δ′相容易在β′相上异质核心长大,在基体中形成复合 δ′/β′相。由于 δ′与 β′相同属于 L12 型晶体结构,原子排列相同,先存在的β′相促进了δ′相的形核,使 δ′相以 β′相为核心,形成 δ′相包裹着核 β′相的复合相δ′/β′。由于复合相界面处的界面电子密度连续性好,使得畸变应力小,又由于中心的β′相具有强的Al—Zr共价键络[7],合金变形时,位错不易切过,核壳结构的复合相 δ′/β′可以有效抑制位错切割,同时提高合金的强度和韧性。
由表5可见,电子密度连续性好的界面,界面能都比较低,与实验结果吻合;电子密度连续性差的界面,界面能都比较高,对应的结合性能较差,伴随着界面弱化,在强外力作用下,易成为出现裂纹萌生或断裂的发源地。
4 结论
1) δ′相与基体之间的界面电子密度在较低的应力下保持连续,δ′相与基体界面的结合强度较大,δ′相的析出起到了界面增强的效果;δ相与基体间界面电子密度在一级近似下不连续,界面折断的共价键数目较多,因此与基体间界面结合较弱,起界面弱化作用。
2) 对于核壳结构的复合相δ′/β′,界面电子密度差较小,其界面能最低。因此,δ′相容易在 β′相上异质核心长大,在基体中形成复合 δ′/β′相,在一定情况下对合金起强化作用。
REFERENCES
[1] LAVERNIA E J, SRIVATSAN T S. Review of strength and fracture behavior and ductility of Al-Li alloys[J]. J Mater Sci,1990, 25: 1137−1142.
[2] JOH C H, YAMADA K, MIURA Y. The coarsening behavior of Al3Li precipitates in Al-Li alloys[J]. Materials Transaction, 1999,40: 439−444.
[3] GABLE B M, ZHU A W. Micorstructural evolution and mechanical properties of a Al-Li-Cu alloy[J]. J Light Metals,2001, 1: 1−14.
[4] NIE J F, MUDDLE B C. The age-hardening response in high strength Al-Li-Cu alloy[J]. Mater Sci Eng A, 2001, 319/321:4448−4455.
[5] CHEN Y L, LI J F. Microstructures and mechanical properties of an Al-Li-Cu-Mg alloy[J]. Mater Sci Forum, 2007, 546/549:995−1002.
[6] 高英俊, 黄创高, 莫其逢. Al-Li合金时效初期的价键分析[J].中国有色金属学报, 2005, 15(7): 1069−1074.GAO Ying-jun, HUANG Chuang-gao, MO Qi-feng. Electronic structure of Al-Li alloy under earlier ageing condition[J]. The Chinese Journal of Nonferrous Metals, 2005, 15(7): 1069−1074.
[7] GAO Ying-jun, MO Qi-feng, CHEN Hua-ning. Atomic bonding and mechanical properties of Al-Li-Zr alloy[J]. Mater Sci Eng A,2009, 499: 299−303.
[8] MONDOLFO L F. Structure and properties of aluminum alloys[M]. London: Butterworths Press, 1976: 100−250.
[9] HOWE J M, LAUGHLIN D E. A high-resolution transmission electron microscopy investigation of the δ′ precipitate structure in an Al-Li alloy[J]. Philosophical Magazine A, 1988, 57(6):955−969.
[10] 张瑞林. 固体与分子经验电子理论[M]. 长春: 吉林科学技术出版社, 1993: 1−250.ZHANG Rui-lin. Empirical electron theory in solids and molecules[M]. Changchun: Jilin Science and Technology Press,1993: 1−250.
[11] 刘志林, 李志林, 刘伟东. 界面电子结构与界面性能[M]. 北京: 科学出版社, 2002: 23−155.LIU Zhi-lin, LI Zhi-lin, LIU Wei-dong. Electron structure and properties of interface[M]. Beijing: Science Press, 2002:23−155.
[12] LIU Zhi-lin, LI Zhi-lin, SUN Zhen-guo. Catalysis mechanism and catalyst design of diamond[J]. Metall Mater Trans A, 1999,30: 2757−2763.
[13] GAO Ying-jun, HOU Xian-hua, MO Qi-feng. Atomic bonding of precipitate and phase transformation of Al-Cu-Mg alloy[J]. J Alloy & Compounds, 2007, 441: 241−245.
[14] 高英俊, 陈华宁, 韦 娜, 文春丽, 黄创高. Al-Mg-Si合金的U1和 U2析出相的原子成键与性能[J]. 中国有色金属学报,2010, 20(7): 1267−1274.GAO Ying-jun, CHEN Hua-ning, WEI Na, WEN Chun-li,HUANG Chuang-gao. Atomic bondings and properties of U1 and U2 phases of Al-Mg-Si alloy[J]. The Chinese Journal of Nonferrous Metals, 2010, 20(7): 1267−1274.
[15] 文春丽. Al-Li-Cu-Mg合金相界面的价电子结构与力学性能[D] .南宁: 广西大学, 2010.WEN Chun-li. Electronic structure of interface boundary and mechanical properties of Al-Li-Cu-Mg alloy[D]. Nanning:Guangxi University, 2010.
[16] BORCHERS C, BORMANN R. Determination of low-temperature inter facial energies from a pair interaction model [J]. Acta Mater, 2005, 53: 3695−3701.
[17] BAUMANN S F, WILLIANS D B. A new method for the determination of the precipitation-matrix interfacial energy[J].Scripta Mater, 1984, 18: 611−616.
[18] HYLAND R W, ASTA M, FOILES S M. Al/Al3Zr interphase boundary energy calculations[J]. Acta Mater, 1998, 46(10):3667−3678.
[19] GAYLE F W, VANDER S. Composite precipitation of Al-Li-Zr alloy[J]. Scripta Mater, 1984, 18: 473−478.
Interface atomic bonding and mechanical properties of Al-Li-Zr alloy
GAO Ying-jun, WEN Chun-li, MO Qi-feng, LUO Zhi-rong, HUANG Chuang-gao
(College of Physics Science and Engineering, Guangxi University, Nanning 530004, China)
The atomic bonding and interface energy between precipitation and matrix of Al-Li-Zr alloy were calculated by using the “Empirical Electronic Theory in Solid” (EET). The result shows that the electronic density in interface between δ′ phase and matrix, is continuous under one order approximation at low stress condition, which gets a good combination in interface between δ′ phase and matrix, and strengthens the interface; the electronic density in the interface between δ phase and matrix is not continuous under one order approximation, which make the interface combination weak. For the complex phase δ′/β′ with core and shell structure, not only the interface electronic density is smaller, but also the interface energy is lower, which results to form a complex δ′/β′ phase through the inhomogeneous nucleation of δ′ phase on β′ particles. It can reveal the reason why the precipitation can strengthen and weaken the alloy and thus affect the alloy properties.
Al-Li-Zr alloys; atomic bonding; interface, mechanical property
TG111
A
1004-0609(2011)09-2202-07
国家自然科学基金资助项目(50661001,50061001);广西自然科学基金资助项目(0991026, 0832029, 0639004);广西研究生教育创新计划资助项目(105931001015,105931003070)
2010-09-25;
2011-01-27
高英俊,教授,博士;电话:0771-3232666;E-mail: gaoyj@gxu.edu.cn
(编辑 何学锋)
