光学纯Communesin F中间体的合成*
2011-11-22黄小平吴昊星
黄小平, 吴昊星, 宋 颢
(四川大学 华西药学院,四川 成都 610041)
吲哚类生物碱Communesin A~Communesin F[1](Ⅰa~Ⅰf)是近年来从海鞘中分离出来的一类结构复杂、骨架新颖的海洋天然产物,具有抗肿瘤和杀虫等重要的生理活性。Ⅰ一经发现即因其新颖的骨架结构和显著的生理活性而备受有机化学家的关注,成为有机化学全合成研究领域的明星分子。然而由于Ⅰ的分子结构复杂,具多环稠和体系及多个手性中心,其合成具有极大的挑战性,近年来秦勇课题组[2]和Weinreb课题组[3,4]成功地完成了Ⅰf消旋体的全合成。结构分析表明,Ⅰf与Ⅰa~Ⅰe最显著的区别在于11-C侧链上的环氧官能基的有无。由Ⅰf侧链上的双键可通过简单的官能基转化完成Ⅰa~Ⅰe侧链环氧环的构建。由此可见,如能顺利完成光学纯Ⅰf的合成,无疑将对光学纯Ⅰa~Ⅰe的合成提供重要参考。
众所周知,在反应过程中引入手性拆分试剂是将消旋体制备成光学纯化合物最直接简单的方法。Ellman[5]于1997年通过固相合成首次得到了物理、化学和光学性质均稳定的新型功能有机小分子光学纯叔丁基亚磺酰胺(Ⅱ)。因其优秀的手性诱导作用以及叔丁基亚磺酰基容易脱去的优点,Ⅱ被广泛应用于不对称合成中。近年来涌现出大量关于Ⅱ手性诱导合成具有光学活性含氮有机中间体的报道[6,7]。光学纯Ⅱ已成为制备具有重要生理活性手性胺类医药中间体的关键手性源。鉴于光学纯Ⅱ在手性诱导中的重要作用,我们在已成功完成Ⅰf消旋体的全合成的基础上,对基于手性拆分的Ⅰf不对称合成开展了研究。
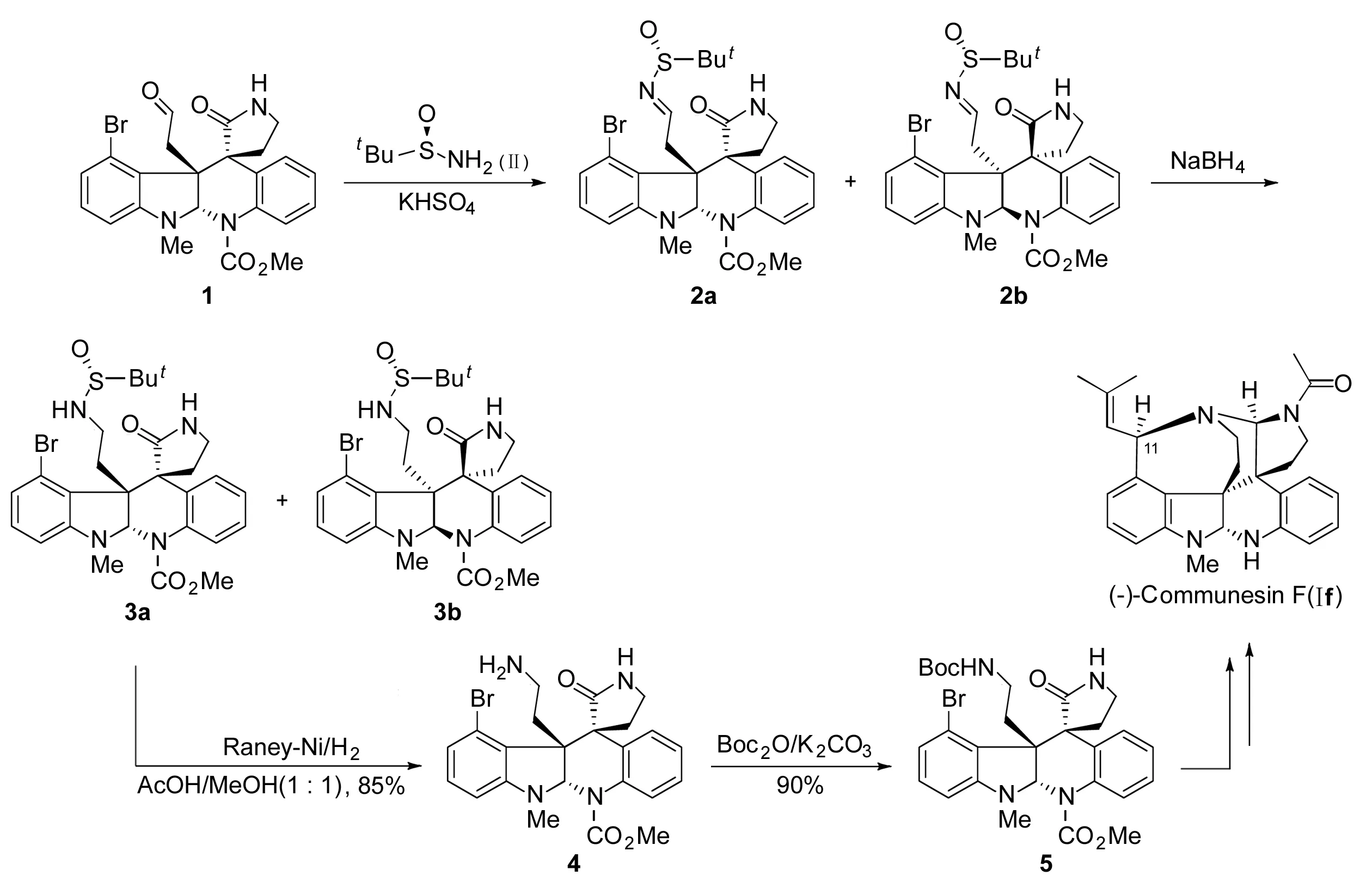
Scheme 1
本文参照文献[8]方法,在KHSO4作用下,1(在Ⅰf消旋体的全合成过程中所获得的醛中间体)首先与光学纯Ⅱ缩合得到希夫碱中间体2a和2b; 2不经纯化,直接在NaBH4的作用下还原,再通过柱层析分离即可得到摩尔比为1 ∶1的非对应异构体3a和3b;3a在雷尼镍的作用下,氢化脱去叔丁基亚磺酰基完成消旋体拆分得光学纯4(Scheme 1)。4的氨基用Boc保护得5(与合成Ⅰf消旋体过程中相同的关键中间体)。5可按文献[2]方法完成光学纯Ⅰf的全合成。
为了进一步确定化合物的绝对立体化学,我们对光学纯3a,3b和5的单晶培养进行了初步尝试,然而令人遗憾的是,在尝试了甲醇、乙醇、异丙醇、乙醚、异丙醚、乙二醇二甲醚、苯等常用结晶溶剂后,均未能如愿获得单晶。
1 实验部分
1.1 仪器与试剂
Varian Unit INOVA 400型高分辨超导核磁共振仪(CDCl3为溶剂,TMS为内标)。1按文献[2]方法自制;其余所用试剂均为市售化学纯或分析纯;溶剂经常规干燥处理。
1.2 合成
(1) 3的合成
在三颈烧瓶中加入1 2.41 g(5 mmol), Ⅱ 605 mg(5 mmol),无水KHSO43.4 g(25 mmol)和甲苯30 mL,搅拌使其溶解,回流反应13 h(TLC跟踪)。过滤,滤液浓缩得2粗品。
将2粗品溶于THF(30 mL)中,搅拌下于0 ℃分批加入NaBH4380 mg(10 mmol),加毕,反应10 min。加入饱和NH4Cl溶液(20 mL)淬灭反应,分液,水相用CH2Cl2(3×50 mL)萃取,合并有机相,用无水NaSO4干燥,减压浓缩,残余物经硅胶柱层析[洗脱剂:A=V(CH2Cl2) ∶V(MeOH)=7 ∶1]纯化得白色固体3[n(3a) ∶n(3b)=1 ∶1]1.4 g,收率93%(以1计算)。
3a:1H NMRδ: 1.21(s, 9H), 1.85~1.93(m, 1H), 2.26~2.32(m, 1H), 2.42~2.46(m, 1H), 2.50(s, 3H), 2.91~3.18(m, 2H), 3.08~3.18(m, 2H), 3.28~3.33(t, 1H), 3.46~3.51(dd, 1H), 3.84(s, 3H), 5.93(d,J=7.6 Hz, 1H), 6.06(br s, 1H), 6.62(d,J=8.0 Hz, 1H), 6.75(t,J=8.0 Hz, 1H), 7.02(t,J=8.2 Hz, 1H), 7.15(t,J=7.0 Hz, 1H), 7.22(br s, 1H), 7.51(d,J=7.6 Hz, 1H), 8.31(s, 1H)。
3b:1H NMRδ: 1.25 (s, 9H), 1.83~1.92(m, 1H), 2.25~2.30(m, 1H), 2.48(s, 3H), 2.91~2.97(dd, 1H), 3.10~3.17(m, 2H), 3.26~3.31(m, 2H), 3.52~3.55(m, 2H), 3.86(s, 3H), 5.97(d,J=7.6 Hz, 1H), 6.10(br s, 1H), 6.68(d,J=7.6 Hz, 1H), 6.79(t,J=8.0 Hz, 1H), 7.04(t,J=8.2 Hz, 1H), 7.18(t,J=8.2 Hz, 1H), 7.24(d,J=7.6 Hz, 1H), 7.35(br s, 1H), 7.45(d,J=7.6, 1H)。
(2) 4的合成
在三颈烧瓶中加入AcOH 5 mL, MeOH 5 mL和31.2 g(2 mmol),搅拌使其溶解后加入雷尼镍58.7 mg(1 mmol),氢气氛(0.1 MPa)下于室温反应20 h(TLC跟踪)。过滤,滤液用CH2Cl2稀释,用1 mol·L-1KOH溶液调至pH 8;分液,有机层用水洗涤,无水NaSO4干燥,减压浓缩,残余物经硅胶柱层析(洗脱剂:A=5 ∶1)纯化得白色固体4 840 mg,产率85%;1H NMRδ: 1.85~1.93(m, 1H, NHCH2CH2), 2.02(br s, 2H, NH2), 2.14~2.21(m, 1H, NHCH2CH2), 2.48(s, 3H, NCH3), 2.61(dt,J=11.6 Hz, 4.4 Hz, 1H, NHCH2CH2), 2.70(dt,J=11.2 Hz, 4.0 Hz, 1H, NHCH2CH2), 2.97(dd,J=14.0 Hz, 7.2 Hz, 1H, NHCH2), 3.06(dt,J=12.0 Hz, 4.8 Hz, 1H, NHCH2), 3.29(t,J=9.4 Hz, 1H, NHCH2), 3.54(dd,J=17.0 Hz, 9.4 Hz, 1H, NHCH2), 3.86(s, 3H, OCH3), 5.97(d,J=7.6 Hz, 1H, ArH), 6.06(br s, 1H, NCHN), 6.67(d,J=8.0 Hz, 1H, ArH), 6.78(t,J=8.0 Hz, 1H, ArH), 6.87(br s, 1H, ArH), 7.05(t,J=8.2 Hz, 1H, ArH), 7.19(t,J=7.0 Hz, 1H, ArH), 7.24(br s, 1H, ArH), 7.45(d,J=8.0 Hz, 1H, NH)。
(3) 5的合成
N2保护,在三颈烧瓶中加入4 484 mg(1 mmol)的CH2Cl2(10 mL)溶液,Na2CO3212 mg(2 mmol)和(Boc)2O 220 mg(1 mmol),搅拌下于室温反应24 h(TLC跟踪)。过滤,滤液真空浓缩,残余物经硅胶柱层析[洗脱剂:V(乙酸乙酯) ∶V(石油醚)=1 ∶1]纯化得白色固体5 502.2 mg,产率90%;1H NMRδ: 1.45(s, 9H, CH3in Boc), 1.87~1.96(m, 1H, NHCH2CH2), 2.24~2.27(br d, 1H, NHCH2CH2), 2.50(s, 3H, NCH3), 2.94(dd,J=14.2 Hz, 7.0 Hz, 2H, NHCH2CH2), 3.04(m, 2H, NHCH2), 3.31(t,J=9.6 Hz, 1H, NHCH2), 3.50(dd,J=16.8 Hz, 5.2 Hz, 1H, NHCH2), 3.88(s, 3H, OCH3), 4.64(br s, 1H, NH), 5.98(d,J=7.6 Hz, 1H, ArH), 6.10(br s, 1H, NH), 6.38(s, 1H, NCHN), 6.68(d,J=7.6 Hz, 1H, ArH), 6.78(t,J=7.8 Hz, 1H, ArH), 7.05(t,J=8.2 Hz, 1H, ArH), 7.20(t,J=7.6 Hz, 1H, ArH), 7.28(s, 1H, ArH), 7.45(d,J=7.6 Hz, 1H, ArH)。
2 结果与讨论
本文以Communesin F消旋体的全合成过程中所获得的醛中间体1为原料,在光学纯叔丁基亚磺酰胺的手性诱导下,通过加成和还原两步反应得到比例为1 ∶1的非对应异构体3a和3b。在酸性条件下,3a的叔丁基亚磺酰胺脱去,用Boc对其氨基进行保护后,得到合成Communesin F消旋体过程中的关键前体化合物5。从光学纯中间体5出发,参照原有的Communesin F消旋体的全合成路线,经Heck反应、PPTs作用下的环合、酰胺键活化、脱Boc保护基、在硅胶作用下的环合、脱甲酸甲酯保护基和亚胺键还原乙酰化等7步反应即可完成光学纯Communesin F的合成。其中非对应异构体3a,3b和5绝对立体化学的确定正在进行当中。
[1] Numata A, Takahashi C, Ito Y,etal. Communesins,cytotoxic metabolites of a fungus isolated from a marine alga[J].Tetrahedron Lett,1993,34:2355-2358.
[2] Yang J, Wu H X, Shen L Q,etal. Total synthesis of (±)-communesin F[J].J Am Chem Soc,2007,129:13794-13795.
[3] Peng Liu, Jae Hong Seo, Steven M Weinreb. Total synthesis of the polycyclic fungal metabolite (±)-communesin F[J].Angew Chem Int Ed,2010,49:2000-2003.
[4] Peng Liu, Jae Hong Seo, Steven M Weinreb. Evolution of a strategy for total synthesis of the marine fungal alkaloid (±)-communesin F[J].J Org Chem,2010,75:2667-2680.
[5] Liu G C, Cogan D A, Ellman J A. Catalytic asymmetric synthesis oft-butanesulfinamide.Application to the Asymmetric Synthesis of Amines[J].J Am Chem Soc,1997,119:9913-9914.
[6] Ellman J A. Applications oft-butanesulfinamide in the asymmetric synthesis of amines[J].Pure Appl Chem,2003,75:39-46.
[7] Ellman J A, Owens T D, Tang T P. Acc.N-t-butanesulfinyl imines:Versatile intermediates for the asymmetric synthesis of amines[J].Chem Res,2002,35:984-995.
[8] Huang Zh Y, Feng P, Wang Y,etal. KHSO4mediated condensation reaction oft-butanesulfinamide with aldehyde.Preparation oft-butanesulfinyl aldimine[J].Synlett,2005:1334-1336.
