新型配合物[Cu(cyclen)(H2O)]2+的密度泛函理论研究
2011-11-20朱海燕任颜卫姚焕英
朱海燕,李 珺,任颜卫,姚焕英
(1渭南师范学院化学与生命科学学院,陕西渭南714000;2西安交通大学材料科学与工程学院,西安710049;3西北大学化学系,西安710069)
新型配合物[Cu(cyclen)(H2O)]2+的密度泛函理论研究
朱海燕1,2,李 珺3,任颜卫3,姚焕英1
(1渭南师范学院化学与生命科学学院,陕西渭南714000;2西安交通大学材料科学与工程学院,西安710049;3西北大学化学系,西安710069)
[Cu(cyclen)(H2O)]2+(cyclen=1,4,7,10-四氮杂环十二烷)是一种新型的大环配合物.在其实验所测晶体结构基础上,文章运用量子化学密度泛函理论中的B3LYP/6-31G(d)方法,对其结构参数进行优化和频率分析,结果表明优化结构为稳定结构,且与实验吻合良好.最后,对配合物的前沿分子轨道及自然电荷布居进行了分析.
1,4,7,10-四氮杂环十二烷;铜配合物;密度泛函理论;量子化学计算
天然水解酶是高效的催化剂,在生物化学反应中起着非常重要的作用[1].在温和条件下,生物体内的各种生物化学反应,几乎都是通过这类酶的催化作用实现的.随着蛋白质、遗传、分子生物和化学生物工程的飞速发展,人类对天然水解酶的需求日益增加.无论从品种上还是数量上来看,天然水解酶都远远不能满足发展的要求,而且这种生化试剂制备困难,价格昂贵.因此,探索合成人工核酸酶已逐渐成为当前生物无机化学十分活跃的前沿领域之一[2].大环多胺类化合物1,4,7,10-四氮杂环十二烷(cyclen)是在冠醚和穴醚之后人们设计合成的一种十分重要的功能型配体[3].由于与血红素、叶绿素等生物活性物质的骨架(卟啉环)结构相似,且具有大环效应和多氮的配位环境,易于同金属形成配合物,因此,过渡金属与cyclen的配合物经常被用来模拟金属核酸酶的活性中心[4-6].我们的作者任颜卫等以cyclen为配体,通过与相同物质量的CuCl2·2H2O在碱性溶液中作用成功地合成了一种新型的配合物[Cu(cyclen)(H2O)]2+(a),并采用单晶衍射分析了其结构[7].本文运用密度泛函(DFT)方法对实验测得配合物(a)的结构进行了量子化学几何优化,在优化结构基础上研究了其前沿轨道排布及自然键轨道布居等.
1 计算方法
Gaussian 03是一个功能强大的量子化学综合软件包,常用于研究许多有关化学领域的课题.其可执行程序可在不同型号的大型计算机、超级计算机、工作站和个人计算机上运行,并相应有不同的版本.Gaussian 03可以用来计算过渡态能量和结构、键和反应能量、分子轨道、原子电荷和电势、振动频率、红外和拉曼光谱、核磁性质、极化率和超极化率、热力学性质、反应路径,计算可以对体系的基态或激发态执行.可以预测周期体系的能量,结构和分子轨道.近年来Gaussian 03程序在配合物电子结构以及性质研究方面应用广泛,且计算结果与实验符合良好.因此为了详尽地研究配合物(a)的结构、电荷排布及轨道布居等,本文用Gaussian 03程序在深腾1800机群上进行计算.
对于有机金属配合物,密度泛函理论(Density Functional Theory,DFT)是一种实用且有效的计算方法[8-10].本研究以X-ray单晶衍射所测晶体结构为基础,采用密度泛函理论的 B3LYP[11-13]方法,全电子基组6-31G(d)优化其结构,并以同水平下的频率计算确认其稳定构型,当所有的频率为正,保证能量的二阶导数矩阵的本征值为正数,即所计算的体系能量对应于势能面上的最小点.最后,对优化所得的分子的稳定几何构型进行前沿轨道(包括HOMO-1,HOMO,LUMO和LUMO+1)排布及自然键轨道(NBO)电荷布居分析.
2 结果与讨论
2.1 结构
图1是化合物a优化分子结构图,表1是化合物的主要键长、键角参数的理论与实验值的比较.可以看出计算与实验所得键长、键角参数基本一致,表明计算与实验所得结构一致.配合物a由1个Cu原子、1个结晶水和1个cyclen大环配体构成.中心配位离子Cu2+与大环配体cyclen中的4个N原子和1个水分子直接配位,形成一个不规则的四角锥体几何构型,cyclen的四个N原子位于四角锥的底面.Cu2+到这个面的距离为0.4516A,配位水分子位于轴向位置,Cu-O键长为2.147A,Cu2+到四个氮原子的距离并不完全相等.计算结果表明,化合物a中的cyclen结构具有一定的几何对称性,但是实验结果表现出与对称结构的偏差,引起这种偏差的原因主要与结晶水分子并不位于轴向的中心,且水分子上的两个氢原子并不一定与轴向对称,而在Gaussian计算中很难识别这一点.值得注意的是,计算和实验的偏差在量子化学计算误差允许范围内,该结构可以正确地反映配合物a的结构信息,因此该优化结构被用于后面配合物其它性质的研究中.
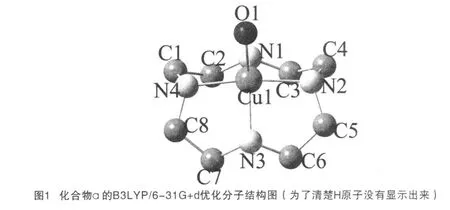
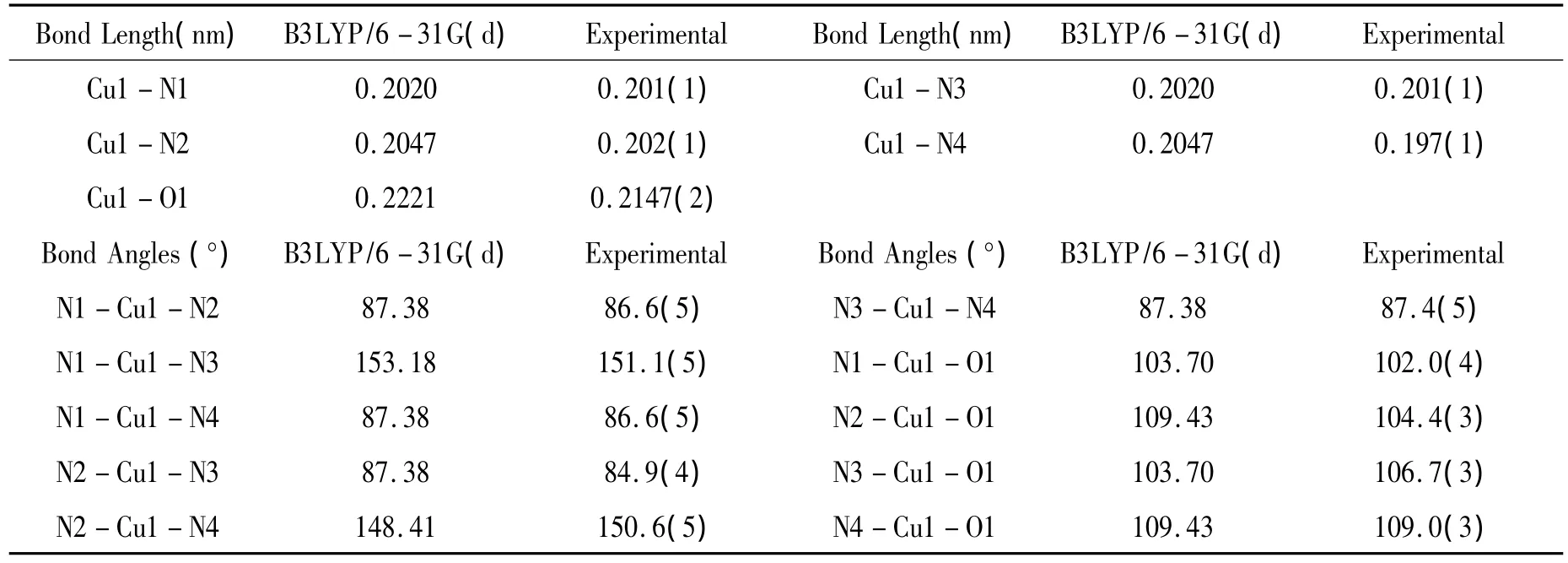
表1 化合物a主要键长(nm)键角(deg)计算值及实验值
2.2 前沿轨道布居
根据分子轨道理论,前沿(线)轨道(HOMO,LUMO)及邻近的分子轨道对化合物的反应活性影响较大,最高占据分子轨道(HOMO)及其邻近的占据轨道具有 优先提供电子的作用,最低空分子轨道(LUMO)及附近空轨道具有接受电子的作用.为探索化合物的电子结构与成键特征,对化合物的分子轨道排布进行分析,对参与组合的所有原子轨道系数的平方和进行归一化,各原子轨道系数平方在轨道系数平方和中的比例表示该部分在分子轨道中的贡献.化合物的结构分析表明,化合物a的分子结构具有一定的晶体学对称性.根据原子所处的环境把化合物a的原子分为6组,分别为:Cu1;O1;N(I)包括N1和N3;N(II)包括N2和N4;C(I)包括C1、C4、C5、和C8;C(II)包括C2、C3、C6和C7.表2列出了化合物的前沿轨道系数分布情况,图2为化合物a的前沿(线)轨道示意图.
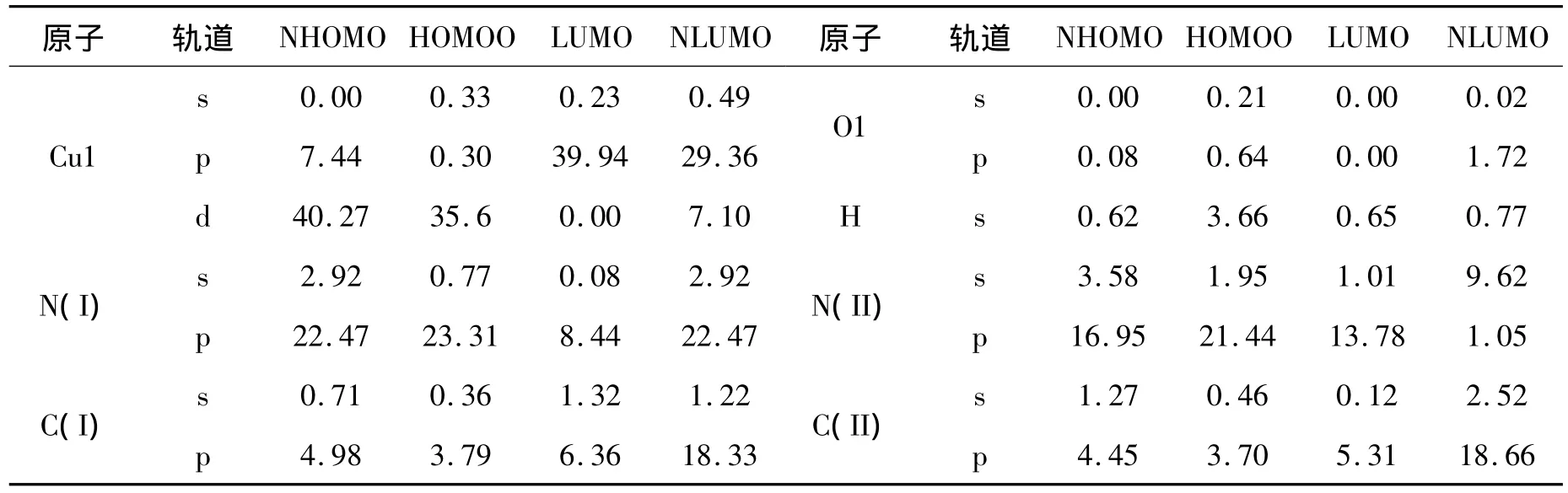
表2 配合物a的前线轨道系数分布(%)
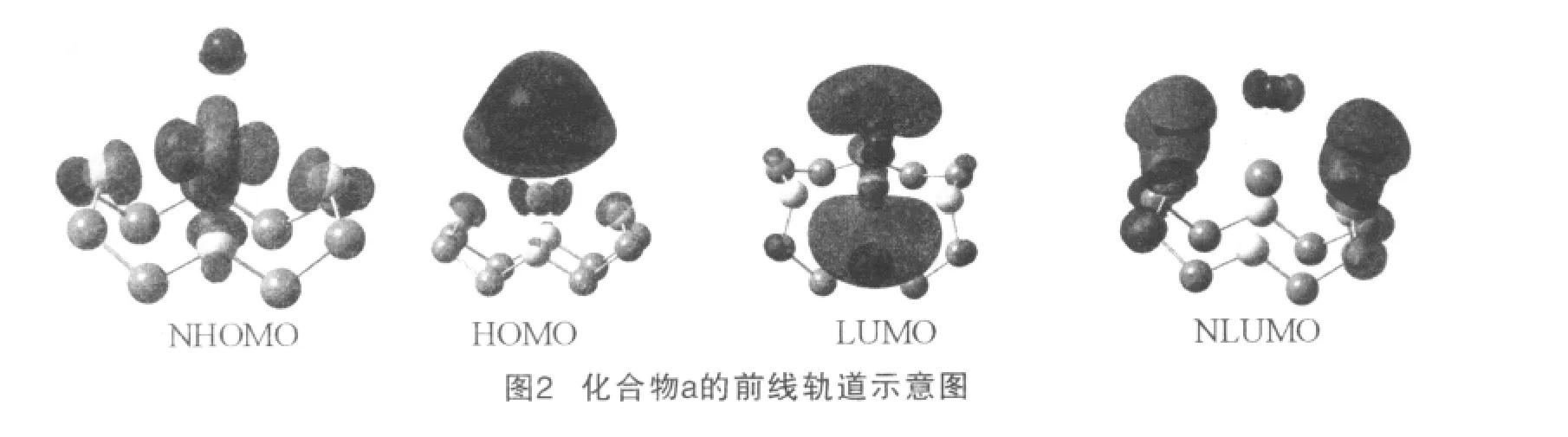
表2和图2表明,化合物a的最高占据轨道(NHOMO和HOMO)主要来自Cu原子的d型轨道,最低非占据轨道(LUMO和NLUMO)主要来自Cu原子的p型轨道.除LUMO外,N(I)对各前沿轨道的贡献略大于N(II),C(I)对前沿轨道的贡献也略大于C(II).H原子以及配位原子O对所有前沿轨道的贡献均较小.因而,标题化合物中N(I)的活性稍高于N(II),C(I)的活性稍高于C(II).Cu原子与配位原子N是化合物的活性部位,展现出丰富的反应性,是化学反应或催化作用的活性区域.cyclen大环状结构使得配合物具有非常大的比表面积,为化学反应和催化作用的发生创造了理想条件.
2.3 自然键轨道(NBO)布居
表3为自然键轨道布居分析(NPA)所得原子电荷分布.从表3可以看出,正电荷主要分布在中心原子Cu上,负电荷主要分布在配位原子O和N上.所有的C原子都带负电,所有氢原子都带正电.N(I)较N(II)所携带负电荷稍多一些.O1约带1 e的负电荷,与O1相连的两个H原子分别携带大约0.5 e的正电荷,整个H2O分子显电中性,因此在形成配合物的过程中没有发生电荷的转移.Cu2+所带电荷为1.1434 e,表明在配合物形成过程中大约有0.86 e的正电荷由Cu2+离子流向cyclen大环.
自然键轨道系数分析显示,Cu-N(I)键的轨道的杂化系数为s0.16p0.84d0.00;Cu-N(II)键的轨道的杂化系数为s0.17p0.83d0.00;Cu与所有N原子的轨道杂化系数几乎相等,其中p形式约为s形式轨道系数的5倍,d形式的轨道没有参与杂化.

表3 原子的自然电荷布居(e)
3 结论
[Cu(cyclen)(H2O)]2+的晶体结构进优化结果显示,Cu2+与大环配体cyclen中的4个N原子和1个水分子配位,形成一个不规则的四角锥体几何构型,4个N原子与中心Cu原子的距离并非完全相等.进一步的NBO和前沿轨道分析显示,与Cu原子距离较近的N(I)比距其较远的N(II)的活性稍高一些.Cu离子与N原子对前沿轨道的贡献远大于其它原子,可能成为化学反应的活性部位或催化作用的敏感区域.cyclen大环结构使配合物具有足够大的比表面积,为化学反应和催化作用的发生创造了必要条件.
[1]杨频,高飞.生物无机化学原理[M].北京:科学出版社,2002.
[2]高飞,阴彩霞,杨频.核酸酶催化磷酸二酯键水解断裂作用的配位化学模拟[J].科学通报,2004,49(15):1471-1483.
[3]Melson G A.Coordination Chemistry of Marocyclic Compounds[M].New York:Plenum,1979.
[4]REN Yan-wei,LI Jun,ZHANG Feng-xing,et al.Crystal structure and characterization of a new mixed-valence manganese(III/IV)complex:[Mn2(cyclen)2(μ-O)2〛(ClO4)3·4H2O[J].Chinese Journal of Chemistry,2005,23(4):418-420.
[5]朱海燕,李珺,任颜卫.1,4,7,10-四氮杂环十二烷配体的夹心型双核 Mn配合物[Mn2(cyclen)2(μ-O)2](ClO4)3·4H2O 的量子化学研究[J].化学研究,2010,21(6):14-17.
[6]Bernhardt P V,Lawrance G A,Comba P,et al.Synthesis,physical properties and x-ray crystal structure of an oxovanadium(IV)complex of the pendant-arm macrocycle,13-dimethyl-1,4,8,11 – tetrazacyclotetradecane-6,13-diamine[J].J Chem Soc,Dalton Trans,1990,(29):2859-2862.
[7]REN Yan-wei,LI Jun,ZHAO Su-min,et al.Synthesis,crystal structures and thermal decomposition of two novel supramolecule complexes[Ni(cyclen)(H2O)2](tpa)and[Cu(cyclen)H2O](tpa)·3H2O(cyclen=1 ,4 ,7 ,10-teraazacyclododecane,tpa=dianion of terephalic acid)[J].Structural Chemistry,2005,16(4):439-444.
[8]Barckholtz T A,Bursten B E.On the possible structures of Mn2(CO)(8):Theoretical support for an unprecedented asymmetric unbridged isomer[J].J.Am.Chem.Soc,1998,(8):1926-1927.
[9]Niu S,Hall M B.Modeling the active sites in metalloenzymes 5.The heterolytic bond cleavage of H2in the[NiFe]hydrogenase of Desulfovibrio gigas by a nucleophilic addition mechanism[J].Inorg.Chem,2000,24(32):6201-6203.
[10]Macchi P,Sironi A.Chemical bonding in transition metal carbonyl clusters:complementary analysis of theoretical and experimental electron densities[J].Coord.Chem.Rev,2003,238:383-412.
[11]Becke A D.A new mixing of Hartree-Fock and local density-functional theories[J].J Chem Phys,1993,98(2):1372-1377.
[12]Lee C,Yang W T,Parr R G.Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density[J].Phys Rev B,1988,37(2):785-789.
[13]Schlegel H B.Optimization of equilibrium geometries and transition structures[J].J Comput Chem,1982,3:214-218.
Quantum Chemical Study of a Novel Complex[Cu(cyclen)(H2O)]2+
ZHU Hai-yan1,2,LI Jun3,REN Yan-wei3,YAO Huan-ying1
(1 School of Chemistry and Chemical Engineering,Weinan Teachers University,Weinan 714000,China;2 State Key Laboratory for Mechanical Behavior of Materials,School of Materials Science and Engineering,Xi’an Jiaotong University,Xi’an 710049,China;3 Department of Chemistry,Shaanxi Key Laboratory of Physico-inorganic Chemistry,Northwest University,Xi’an 710069,China)
[Cu(cyclen)(H2O)]2+(cyclen=1,4,7,10-tetraazacyclododecan)is a new type big ring cuprum complex.Based on the crystal structure,its electronic structure and frequency were then calculated by conducting density functional theory within B3LYP/6-the 31G(d)method of quantum chemical calculations,and the results have shown that the optimized structure is stable and is agreement with the experimental results.Finally,the frontier molecular orbital and natural charge population was analyzed.
1,4,7,10-tetraazacyclododecan;cuprum complex;DFT;quantum chemical calculations
O741
A
1009—5128(2011)06—0059—04
2011—04—07
陕西省教育厅专项基金资助项目(2010JK548);渭南师范学院科研计划项目(07YKZ004)
朱海燕(1978—),女,陕西渭南人,渭南师范学院化学与生命科学学院讲师,西安交通大学在读博士.研究方向:储氢材料的计算研究.
【责任编辑 曹 静】
